Trends Sci. 2026; 23(10): 13006
Structure Guided Natural Product Discovery: Isolation and Computational Optimization of Novel Baeckenone-Derived MurA Enzyme Inhibitors
Ilham Kurniawan1, Desmila Idola1,*, Ippei Niwata2, Aulia Umi Rohmatika1,
Hisashi Muramatsu2, Chul-Sa Kim2, Takehiro Kashiwagi2, I Made Artika3,
Wien Kusharyoto4 and Fachrur Rizal Mahendra5
1Faculty of Medicine, Universitas Pembangunan Nasional Veteran Jawa Timur, Surabaya 60294, Indonesia
2Department of Agricultural Chemistry, Faculty of Agriculture and Marine Sciences, Kochi University,
Kochi 7808520, Japan
3Department of Biochemistry, Faculty of Mathematics and Natural Sciences, IPB University,
West Java 16680, Indonesia
4National Research and Innovation Agency, Jakarta Pusat 10340, Indonesia
5Bioinformatic Research Center, Indonesian Institute of Bioinformatics, Jawa Timur 65162, Indonesia
(*Corresponding author’s e-mail: [email protected])
Received: 12 December 2025, Revised: 22 February 2026, Accepted: 1 March 2026, Published: 20 April 2026
Abstract
The rapid spread of multidrug-resistant bacteria has created an urgent, global need for novel antibacterial agents capable of overcoming existing resistance mechanisms. This computational study employs an integrated in silico approach to explore the potential of baeckenone compounds as MurA enzyme inhibitor. We report the isolation of 3 compounds from Baeckea frutescens: (5,7-dihydroxy-8-(1-(2-hydroxy- 4-methoxy-3,3,5-trimethyl -6-oxocyclohexa-1,4-dien-1-yl)-2-methylpropyl)-6-methyl-2-phenylchroman-4-one), Baeckenone B, and Baeckenone L (a novel phoroglucinol). These natural compounds served as starting points for in silico structure-based optimization to predict enhanced MurA binding. All derivatives were evaluated for predicted drug likeness, bioactivity scores, and toxicity profile. Molecular docking against MurA was validated by successful re-docking of fosfomycin (RMSD = 0.394 Å). Binding stability was further assessed through 20 ns molecular dynamics (MD) simulations and MM-PBSA free energy calculations. Structural optimization predicted improved drug-likeness (all modified derivatives complied with Lipinski’s Rule of Five), enhanced bioactivity scores, and suggested in silico membrane integrity antagonist potential without predicted toxicity. Docking analysis indicated higher predicted binding affinities for modified compounds (ΔG = ‒7.3 to ‒7.7 kcal/mol), surpassing the non-covalent binding component of fosfomycin (ΔG = ‒4.5 kcal/mol). MD simulations confirmed enhanced complex stability, with modified ligands exhibiting lower RMSD (0.612 - 1.120 Å), reduced residue fluctuations (RMSF), and favorable MM-PBSA binding energies. Modified Baeckenone B showed the most promising predicted profile (ΔG = ‒7.4 kcal/mol, Ki = 0.58 µM), and stable binding (RMSD = 0.612 Å). This computational study provides a framework for prioritizing baeckenone derivatives for future experimental validation as novel antibacterial agents against multidrug-resistant pathogens.
Keywords: Antibacterial, Baeckenones, Molecular dynamics, Multidrug-Resistant bacteria, MurA inhibitor
Introduction
The rapid escalation of antibiotic resistance poses a critical threat to global health, with multidrug-resistant (MDR) bacterial strains rendering many conventional
therapeutics ineffective [1,2]. This crisis necessitates the exploration of novel antibacterial targets and strategies. The MurA (UDP-N-acetylglucosamine enolpyruvyl transferase) enzyme, which catalyzes the first committed step in bacterial peptidoglycan biosynthesis, represents a promising and validated target [3]. Unlike many existing antibiotics, inhibitors targeting this essential pathway could circumvent common resistance mechanisms, offering a new approach against MDR pathogens.
Natural products remain a vital source for novel drug discovery, particularly for antimicrobial agents [4]. Plants of the genus Baeckea, specifically Baeckea frutescens L., have been traditionally used and studied for their bioactive potential [5]. Phytochemical investigations have revealed that B. frutescens produces specialized metabolites, primarily phloroglucinol derivatives known as baeckenones [6-9]. These compounds have demonstrated various pharmacological activities, suggesting their potential as leads for antibacterial development [5,6]. However, the potential of baeckenones as inhibitors of the MurA enzyme remains largely unexplored. While fosfomycin is a clinically used covalent MurA inhibitor, resistance can arise through mutations in the catalytic cysteine (Cys115) or through fosfomycin-modifying enzymes [10,11]. Non-covalent inhibitors could offer an alternative strategy to overcome such resistance mechanisms. Previous studies on MurA inhibitors have focused on substrate analogs or synthetic compounds [12-14], but comprehensive structure-based optimization of natural phloroglucinol scaffolds for enhanced MurA binding has not been systematically reported. Furthermore, although several baeckenones have been isolated from B. frutescens [7-9], their binding mechanisms, structure-activity relationships (SAR) against MurA, and potential for optimization are unknown.
The utilization of Baeckea frutescens in modern pharmacology reflects a broader trend of integrating ethnomedicinal knowledge with contemporary scientific research. Previous phytochemical studies have identified bioactive compound Baeckea frutescens, prompting our investigation of its baeckenone derivates as potential MurA inhibitors [6]. By harnessing the antibacterial activity of these compounds, researchers aim to develop novel formulations that could enhance the efficacy of existing antibiotics or offer alternative treatments for infections that pose significant challenges to current therapeutic options. In light of the rising threat of antibiotic resistance, this study aims to investigate the potential of Baeckenone extracted from Baeckea frutescens as an inhibitor of MurA. The primary objective of this research is to explore the ability of these flavonoids to inhibit MurA, a crucial enzyme in bacterial cell wall biosynthesis. Therefore, this study aims to bridge these gaps through an integrated approach combining phytochemistry and computational methods. The specific novelty of this study lies in: (i) the first reported isolation of Baekenone L from B. frutescens; (ii) systematic structure-based modification rules applied to the baekenone scaffold (hydroxyl group optimization, steric bulk reduction, and lipophilicity modulation) specifically targeting MurA; and (iii) integrated computational prioritization combining docking, MD simulations, and MM-PBSA to identify modified Baekenone B as a lead candidate requiring experimental validation. This work is a computational discovery study, aiming to identify and prioritize the most promising baeckenone-derived leads for subsequent experimental validation as novel non-covalent MurA inhibitors.
Materials and methods
Equipment and materials
This study employed structure-based optimization using structure activity relationship (SAR) and molecular dynamics (MD) simulation as part of a computational approach. The simulations were performed on a computer system equipped with an Intel Core i9-12900KF processor, 64 GB RAM, and NVIDIA GeForce RTX 4070 Graphics Card, running Windows 11 Professional 64-bit as the primary operating system. The software tools utilized in this research included Yet Another Scientific Artificial Reality Application (YASARA Structure), Molecular Operating Environment (MOE) 2019.0102, UCSF Chimera 1.17.3, Discovery Studio 3.5 Client 2025, Avogadro, and Open Babel GUI.
The 3D structures of the molecules studied,’osfomying the MurA enzyme and ligands, were retrieved from various sources. The MurA enzyme structure (PDB code 1UAE) was obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (https://www.rcsb.org/). The ligand structures drawing from isolation results from Baeckea frutescens including 1) 5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa -1,4-dien-1-yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one, 2) Baeckenone B, and 3) Novel Compound from isolation Baeckenone L, and 4) ‘osfomycin as co-crystallized ligand, were sourced from the PubChem Compound and Substance Database (https://pubchem.ncbi.nlm.nih.gov/), provided by the National Center for Biotechnology Information (NCBI). The 3D structure files were in PDB and PDBQT formats, suitable for molecular docking and dynamics simulations.
Methods
Isolation of novel baeckenone and its derivatives from Baeckea frutescens
B. frutescens leaves were dried at room temperature and macerated with methanol for 24 h. The separation steps of crude extract were: (1) liquid-liquid partition into hexane and water layers, (2) hexane layer was collected and separated using silica open column chromatography into six partitions (100% hexane, 10% ethyl acetate/hexane, 30% ethyl acetate/hexane, 70% ethyl acetate/hexane, 100% ethyl acetate, and 100% methanol), and (3) 10% ethyl acetate/hexane partition was analyzed using GC (SHIMADZU GC system column; HP-5, length 30 m, diameter 0.320 mm, film thickness 0.25 μm, injection injection temperature at 150 °C, and detector temperature at 300 °C) and further separation was conducted by using HPLC (SHIMADZU HPLC system; pump: LC-6AD, column oven: CTO-20A, auto injector: SIL-20AD, detector: SPD-M10A). This HPLC separation was conducted into 2 steps using 2 column HPLC that were silica column; size 10ID×250 mm, type waters and ODS column C-18 UG80; size 4.6 mm ID×250 mm. These steps yielded four compounds and were analyzed using LCMS (SHIMADZU LCMS system, pump: LC-6AD, column oven: CTO-20A, auto-injector: SIL-20AD, detector: SPD-M10A, ODS column C-18 UG80; size 2.0 mm ID×150 mm). The compound structures were measured with a Jeol JNM-ECX500.
Structure based optimization of novel baeckenone and its derivatives
The three isolated baeckenones served as starting scaffolds for in silico optimization targeting the MurA active site. Initial molecular models were constructed using ChemDraw 22.0 (PerkinElmer) and converted to 3D structures, which were then geometry-optimized using the MMFF94 force field in Avogadro 1.2.0. The optimization strategy was guided by analysis of the binding poses of the unmodified baeckenones within the MurA active site (PDB: 1UAE). Key interaction patterns and steric clashes were identified using UCSF Chimera 1.16. Based on this analysis, three optimization principles were applied: (1) Reduction of molecular weight by removing non-essential bulky substituents (e.g., trimethyl groups) while maintaining the core pharmacophore; (2) Strategic introduction of hydrogen bond donors/acceptors at positions predicted to interact with key residues (Cys115, Asp305, Arg120); and (3) Modulation of lipophilicity (LogP) by converting methoxy groups to hydroxyls or vice versa to balance membrane permeability and solubility. The optimization was carried out by applying a combination of the Autodock Tool and UCSF Chimera to refine the geometry and improve the binding affinity of the baeckenones to the MurA enzyme.
Drug-Like parameter analysis
The chemical structures of the baeckenone derivatives extracted from Baeckea frutescens were first prepared in SMILES or 3D file formats. These structures were then analyzed using computational tools such as https://www.molinspiration.com/cgi/properties to evaluate their drug-like properties. Parameters such as lipophilicity (LogP), molecular weight (Mw), hydrogen bond donors/acceptors, and polar surface area (PSA) were assessed, alongside compliance with Lipinskiʼs Rule of 5 and other relevant criteria. Predicted drug-likeness was assessed via the Molinspiration server. Compounds were considered to have “good” predicted drug-likeness if they complied with Lipinskiʼs Rule of Five (MW ≤ 500, LogP ≤ 5, HBD ≤ 5, HBA ≤ 10) and had a topological polar surface area (TPSA) < 140 Ų.
Bioactivity prediction and toxicity evaluation
To predict the bioactivity and toxicity of the selected baeckenone derivatives, computational tools such as PASS (Prediction of Activity Spectra for Substances) and PkCSM (Estimation of Toxic Hazard A Decision Tree Approach) were employed. PASS provides predictions for the biological activity of compounds against a wide range of biomolecules. To predict the biological activity of these compounds, the SEA (Similarity Ensemble Approach) or Pred-activity databases were utilized. These tools predict the potential activity spectra of the compounds across various biological targets, including antimicrobial activity, thus identifying the most promising candidates based on their activity profiles. Bioactivity scores for “antibacterial” and “membrane integrity antagonist” targets were obtained from the same server; scores > 0.5 indicate probable activity, scores between 0 and 0.5 indicate moderate activity, and scores < 0 indicate inactivity. PkCSM evaluates the toxicity potential, including mutagenic, carcinogenic, and organotoxic effects. These tools allow the prioritization of compounds with a favorable therapeutic profile and acceptable toxicity levels. After bioactivity and toxicity predictions, the compounds that demonstrated promising bioactivity and low toxicity were selected for further molecular docking studies.
Molecular docking assessment and visualization of interaction
Protein Preparation: The MurA structure (PDB: 1UAE) was prepared in AutoDockTools by removing water molecules, adding polar hydrogens, and assigning Gaisteiger charges. Ligand Preparation: Ligand structures were energy-minimized using UFF in Avogadro and converted to PDBQT format. Docking Parameters: A grid box of dimensions 18×20×20 Å3 was centered on the fosfomycin binding site (center_x = 38.591, center_y = 24.496, center_z = 43.646). The exhaustiveness was set to 64. For each compound, 20 independent docking runs were performed. Validation: The protocol was validated by re-docking the co-crystallized fosfomycin ligand. The root-mean-square deviation (RMSD) between the docked and crystal poses was 0.394 Å, confirming the reliability of the docking parameters. The docking results were analyzed based on the binding affinity scores and binding energy, with lower values indicating stronger interactions between the ligand and the receptor.
The results of the molecular docking studies were visualized using Discovery Studio Visualization 2025 to examine the interactions between the baeckenone derivatives and the MurA enzyme at the molecular level. These visualization tools provided a clear depiction of key interactions, such as hydrogen bonds, hydrophobic interactions, and electrostatic interactions. The specific residues of the enzyme involved in ligand binding were identified and analyzed to better understand the binding mechanism. The 3D structures of the ligand-enzyme complexes were also visualized to assess how well the baeckenone derivatives fit into the enzymeʼs active site, further validating the docking results. These interaction maps help to determine the potential of the compounds as inhibitors of MurA and their suitability for further experimental validation. The docking study was performed with the MurA enzyme, with the Autodock Vina and Molecular Operating Environment (MOE) used to predict the best binding poses of the baeckenone derivatives. The resulting docking scores were analyzed to select the most promising candidates for further optimization. Statistical comparison of docking scores between unmodified and modified derivatives, as well as with fosfomycin, was performed as described in the Statistical Analysis subsection.
Molecular dynamics simulation
Molecular dynamics (MD) simulations were performed to investigate the binding stability and dynamic behavior of the baeckenone derivatives in complex with MurA. The optimized baeckenone derivatives were docked into the active site of the MurA enzyme (PDB ID: 1UAE). The initial structure of MurA was retrieved from the Protein Data Bank and prepared by removing water molecules and adding hydrogen atoms using YASARA Structure. MD simulations were carried out using YASARA with the AMBER14 force field. Energy minimization was performed to remove steric clashes, followed by a 20 ns simulation under NVT and NPT ensembles. The temperature was maintained at 298 K and pressure at 1 bar. Each system was solvated in a truncated octahedral box of TIP3P water molecules extending 10 Å from the protein surface, and sodium/chloride ions were added to neutralize and achieve a physiological salt concentration of 0.15 M, resulting in approximately 45,000 - 50,000 atoms per simulation box.
To ensure that all analyses reflect equilibrium behavior, the first 2 ns of each trajectory were discarded as equilibration and excluded from production analyses. Convergence was assessed by monitoring the root mean square deviation (RMSD) of protein backbone atoms; systems were considered equilibrated when RMSD fluctuations stabilized (typically after 3 ns). All production analyses including RMSD, root mean square fluctuation (RMSF), radius of gyration (Rg), and MM-PBSA binding free energy calculations were performed on the equilibrated portion of the trajectories (3-20 ns).
Trajectory analysis was performed using YASARA macros md_analyze, md_analyzebindenergy, and md_analyzeres to evaluate complex stability. Key parameters such as RMSD, RMSF, and MM-PBSA energy were calculated to assess dynamic behavior and binding affinity. All computational results were analyzed using Microsoft Excel. Statistical analysis of RMSD was performed as described in the Statistical Analysis subsection; for MD data, descriptive statistics (mean ± SD) are reported.
MM-PBSA binding free energy calculation
The binding free energy (ΔGbind) for each complex was calculated using the Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) method implemented in the macro md_analyzebindenergy of AMBER 20. For each trajectories, 500 snapshots were extracted at regular intervals (every 200 ps) from the equilibrated portion (3 - 20 ns) after confirming system equilibration.
The binding free energy was calculated using the following Eq. (1)
ΔGbind = Gcomplex - (Gprotein + Gligand) (1)
where G represents the free energy of each component, calculated as:
G = EMM + Gsolvation ‒ TS (2)
EMM = Ebonded + Enon-bonded (van der Waals + electrostatic) (3)
Gsolvation = Gpolar + Gnon-polar (4)
The polar solvation energy (Gpolar) was calculated using the Poisson-Boltzmann (PB) equation with the internal dielectric constant set to 1 and the external solvent dielectric constant set to 80. The non-polar solvation energy (Gnon-polar) was estimated based on the solvent-accessible surface area (SASA) using a probe radius of 1.4 Å. The entropic contribution (-TS) was not included in the reported ΔGbind values due to the high computational cost and known limitations of normal mode analysis for such large, flexible systems; thus, the reported values represent enthalpy-dominated binding free energies. All energy components (van der Waals, electrostatic, polar solvation, non-polar solvation) were recorded and analyzed separately to understand the driving forces behind binding. Descriptive statistics (mean ± SD) for MM-PBSA energies are reported
Statistical analysis
All quantitative results from molecular docking are reported as mean ± standard deviation (SD) from 20 independent runs per compound (n = 20). Normality of data distribution was assessed using the Shapiro-Wilk test. For comparisons between 2 groups (e.g., unmodified vs. modified derivatives, or modified derivatives vs. fosfomycin), an independent two-tailed t-test was used when data were normally distributed; otherwise, the Mann-Whitney U test was applied. For multiple comparisons across several compounds, Bonferroni correction was applied by adjusting the significance level to α/n (where n is the number of comparisons). The threshold for statistical significance was set at p < 0.05. The positive control for docking validation was the co-crystallized ligand fosfomycin.
For MD simulations, data from the equilibrated portion (ns 3 - 20) of a single trajectory per complex were used to calculate the mean and SD as measures of temporal fluctuation. Statistical comparisons between groups were not performed for MD data due to the absence of replicate trajectories; instead, differences are discussed descriptively. All statistical analyses for docking were performed using Microsoft Excel and SPSS version 25.
Results and discussion
Isolation and structure elucidation of novel baeckenone and its derivatives from Baeckea frutescens
The methanol crude extract of B. frutescens has shown antimycobacterial activity against Mycobacterium smegmatis JCM6386T, therefore, the separation guided bioassay was conducted to characterize the antimycobacterial compounds. The main partition of B. frutescens leaves containing the antimycobacterial compounds was identified as fraction C obtained from elution with 10% ethyl acetate in hexane, which appeared as a prominent and well-defined peak in the initial HPLC profile (Figure 1(a)). Despite its apparent dominance, further analysis of fraction C by GC (Figure 1(b)) revealed that it was not a single peak but rather a mixture comprising four major compounds along with impurity, as indicated by several sharp and distinct peaks at different retention times.
To achieve better isolation for each compound, fraction C was subjected to further purification using reverse-phase HPLC on an ODS column into two steps using different conditions as described in the method. As shown in Figure 1 (c), this approach separated fraction C into five subfractions (Ca-Ce). Among these, subfraction Cd was selected for additional purification due to its distinct profile. A second stage HPLC separation of Cd resulted in four discrete peaks (Cd-1 to Cd-4) (Figure 1(d)), corresponding to four compounds which were identified as the antimycobacterial compounds present in B. frutescens.
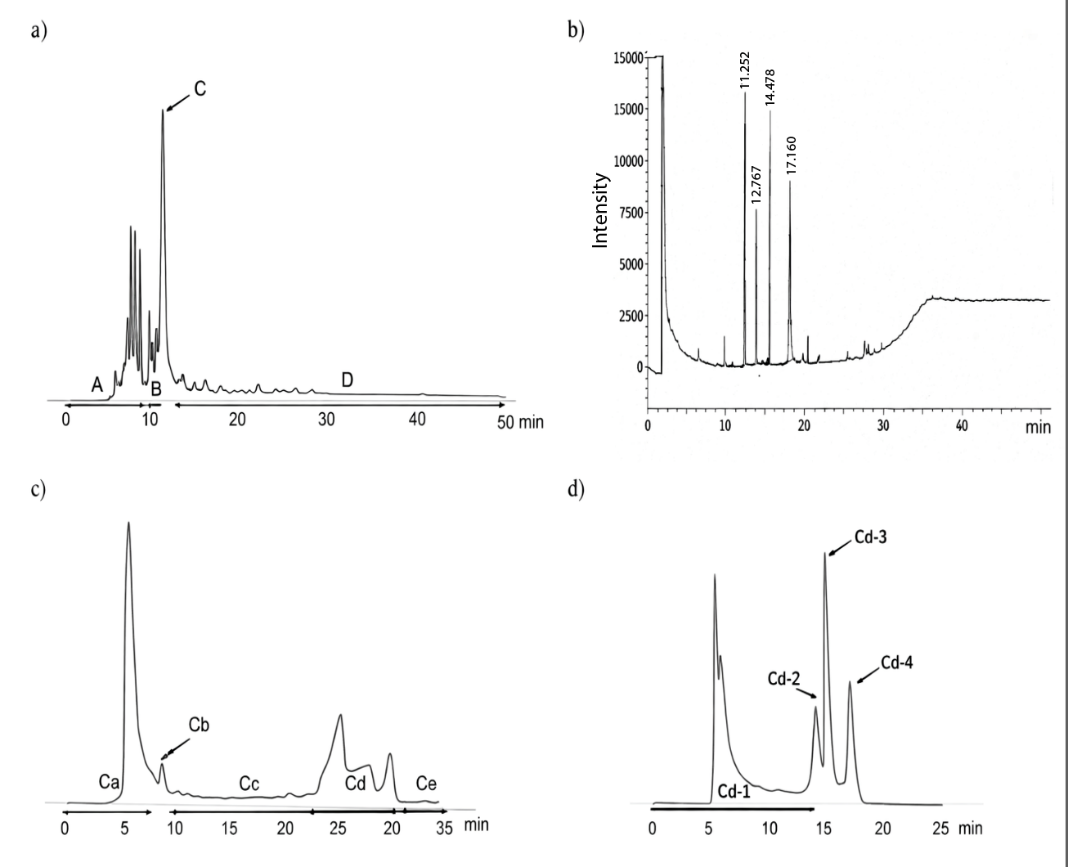
Figure 1 HPLC and GC profile. (a) HPLC profile of 10% ethyl acetate in hexane using silica column, (b) GC profile of fraction C, (c) HPLC profile of fraction C using ODS column, and (d) HPLC profile of fraction Cd using ODS column.
The molecular weights of purified compounds obtained from subfractions Fr. Cd-1, Fr. Cd-2, Fr. Cd-3, and Fr. Cd-4 were determined using LC-MS analysis, as presented in Figure 2. Each fraction exhibited a distinct total ion chromatogram with well-defined peaks, indicating a high degree of purity following the final stage of HPLC separation. The LC-MS spectra at [M+H]+ of these compounds (Fr. Cd-1, Fr. Cd-2, Fr. Cd-3, and Fr. Cd-4 fractions) are m/z 447.35, 507.30, 447.35, and 461.35, respectively. Notably, the identical m/z values observed for Fr. Cd-1 and Fr. Cd-3 suggest that these two fractions may contain different structural isomers.
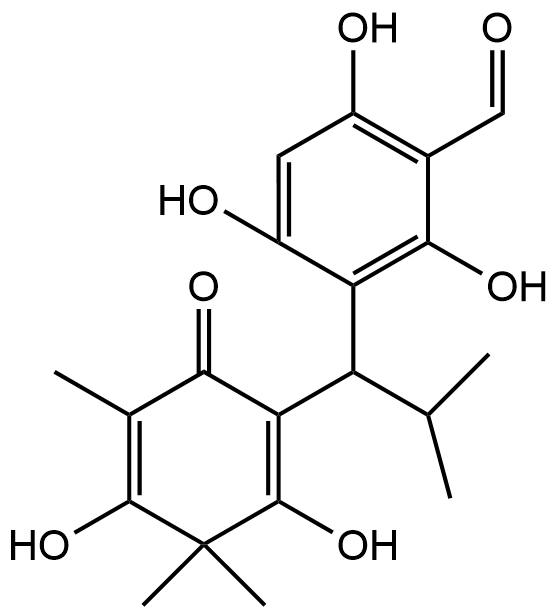
Moreover, NMR analysis results (Table 1, Supplementary data) showed that Fr. Cd-2 is 5,7-dihydroxy-8-(1-(2-hydroxy-4-methoxy-3,3,5-trimethyl-6-oxocyclohexa-1,4-dien-1-yl)-2-methylpropyl)-6-methyl-2-phenylchroman-4-one [7] and Fr. Cd-3 is baeckenone B [8] have been reported, and Fr. Cd-4 is a novel compound while Fr. Cd-1 showed unclear NMR peaks. Compound Cd-4 is 1) obtained as yellow oil, 2) the molecular formula is C26H36O7, 3) IUPAC name is 3-hydroxy-5-methoxy-4,4,6-trimethyl-2-(2-methyl-1-(2,4,6-trihydroxy-3-isobutyryl-5-methylphenyl)butyl)cyclohexa-2,5-dien-1-one, that was determined by chemdrawdirect.perkinelmer.cloud, 4) Fr. Cd-4 possessed a similar structure to baeckenone B or Fr. Cd-3 with additional -CH3 attached at C-10. Further analysis of COSY and HMBC (Figure 3) showed the position of this -CH3 around C-10, C-9, and C-8, and 5) determined as a new phloroglucinol. Since a group of phloroglucinols has been only reported as baeckenone A-K isolated from the leaves of this plant [9], Fr. Cd-4 is named baeckenone L.
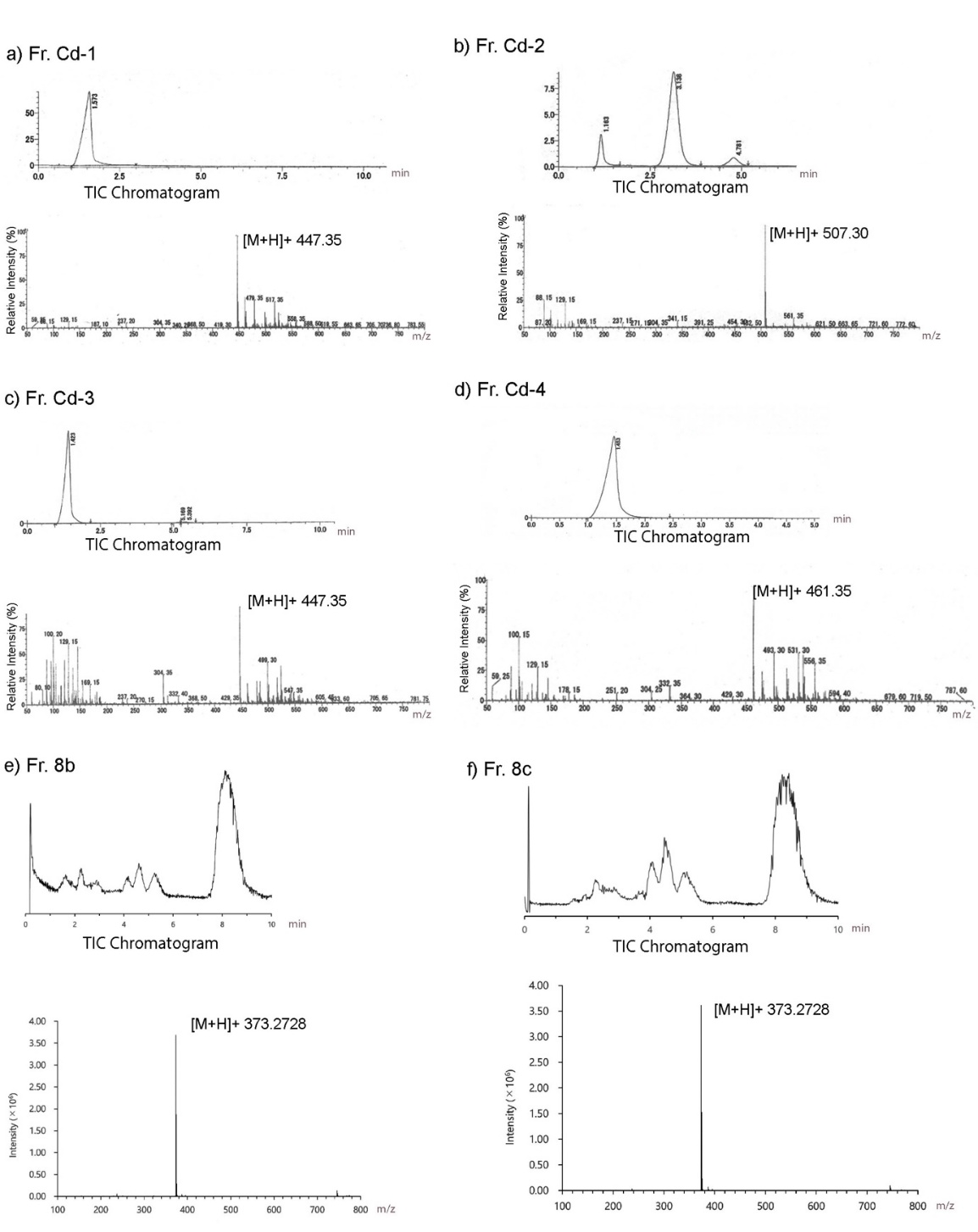
Figure 2 LCMS profile. (a) Fr. Cd-1, (b) Fr. Cd-2 (c) Fr. Cd-3, (d) Fr. Cd-4.
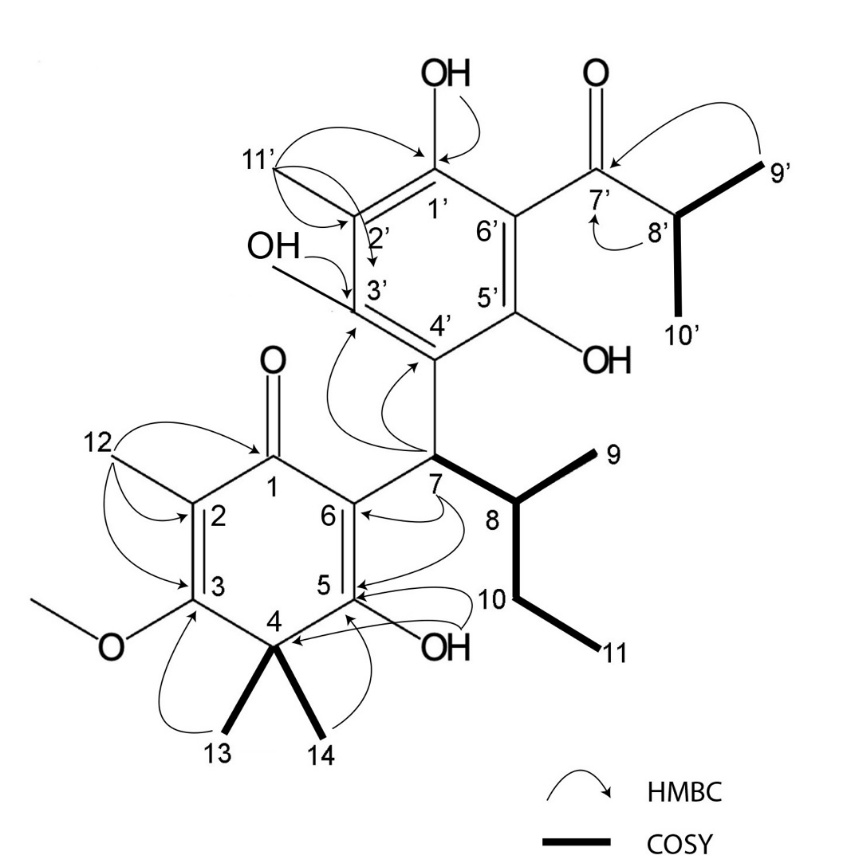
Figure 3 COSY and HMBC of Fr. Cd-4.
Structure based optimization of novel baeckenone and its derivatives
Structure based optimization of Baeckenone derivatives was performed to improve their antibacterial potential by enhancing the molecular features responsible for stable and selective interaction with the target enzyme. The original structures of Baeckenone B and Novel Baeckenone L possess a characteristic chroman-4-one core decorated with multiple hydroxyl groups, which contribute significantly to hydrogen-bond formation and intrinsic bioactivity. However, the presence of sterically bulky substituents and limited polar anchoring sites in certain regions of the molecules may restrict optimal positioning within the receptor’s active pocket. To address these limitations, structural modifications were introduced, focusing on increasing hydrogen-bond donors/acceptors and optimizing the spatial arrangement of hydrophobic fragments in Table 1.
The optimized structures exhibit a more refined interaction profile by selectively enhancing the number and accessibility of hydroxyl groups, particularly on the A- and C-rings. These additional polar groups are positioned to strengthen directional hydrogen-bonding interactions with key catalytic residues, improving the stability of the ligand-receptor complex. Simultaneously, the reduction or removal of large aromatic substituents minimizes steric clashes and allows the modified ligands to adopt deeper and more energetically favorable orientations within the active site. These changes also lower the desolvation energy required for binding, thereby improving ligand efficiency and overall binding affinity. Hydrophobic side chains, especially those retained on the chromanone skeleton, continue to play a critical role in enabling van der Waals interactions within the lipophilic pockets of the bacterial target. By maintaining these hydrophobic contacts while increasing polar complementarity, the optimized derivatives achieve a balanced physicochemical profile that enhances both target engagement and potential membrane permeability. This balance is essential for antibacterial activity, as compounds must efficiently interact with intracellular enzymatic targets while retaining adequate passive diffusion capability. Overall, the structural refinements introduced into Novel Baeckenone L and its related analogs result in improved molecular complementarity, enhanced binding capability, and greater predicted antibacterial potency. These modifications provide mechanistic insight into the structure activity relationship (SAR) of the Baeckenone scaffold and highlight the potential of the optimized derivatives as promising antibacterial candidates for further biochemical and cellular evaluation.
Table 1 Structure-based optimization of novel baeckenone and its derivatives.
Compounds |
Experimental/Original structure |
Modified/Optimization structure |
Fraction Cd-2; 5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4-dien -1-yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one |
|
|
Fraction Cd-3; Baeckenone B |
|
|
Fraction Cd-4; Baeckenon L |
|
|
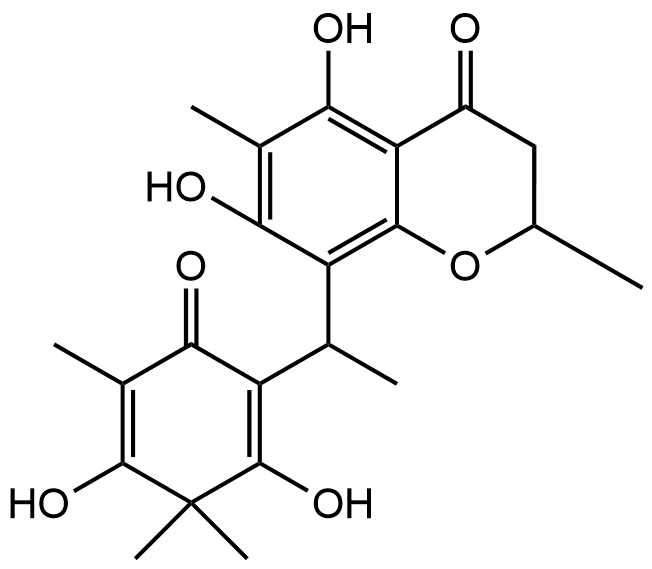
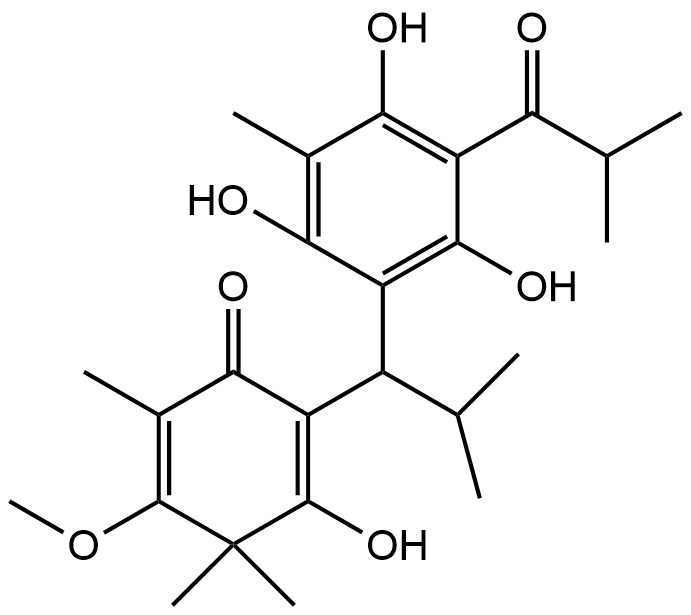
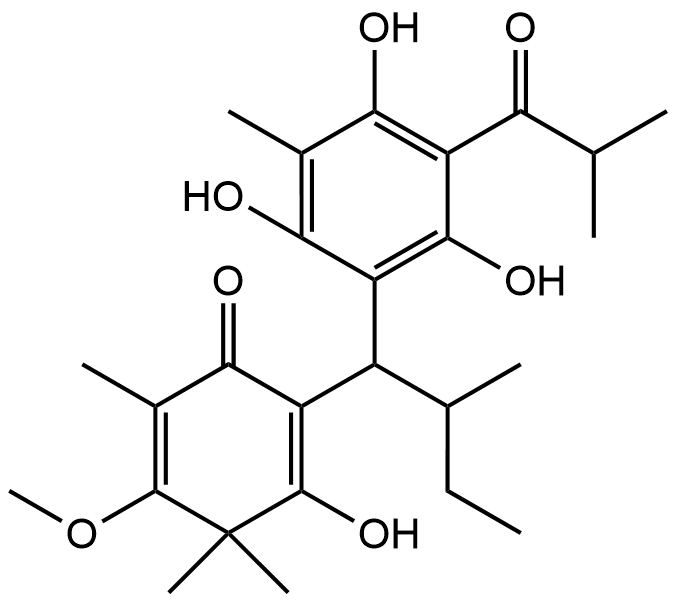
Recent advancements in the structural optimization of Baeckenone derivatives underscore their potential as novel antibacterial agents, particularly through interactions with the MurA enzyme, a critical target in bacterial cell wall synthesis. The chroman-4-one scaffold, which forms the core of Baeckenone B and Novel Baeckenone L, contains functional hydroxyl groups that facilitate hydrogen-bonding interactions, essential for effective enzyme binding [13,14]. However, the original structures of these derivatives feature bulky substituents and limited polar anchoring sites, which can hinder optimal receptor engagement. To address these challenges, structural modifications were introduced to enhance the hydrogen-bonding capabilities and reduce steric hindrance, particularly through the strategic placement of hydroxyl groups on the A- and C-rings [15]. These modifications aim to improve the positioning of the derivatives within the active site of MurA, minimizing steric clashes and desolvation energy, which in turn optimizes binding efficiency and affinity [16]. By fine-tuning the number and accessibility of hydroxyl groups, the modified derivatives enhance hydrogen-bonding interactions with critical catalytic residues, thereby improving binding stability. Additionally, the optimization of hydrophobic side chains plays a crucial role in enhancing the van der Waals interactions between the ligands and the lipophilic pockets of the MurA enzyme. These hydrophobic interactions, combined with the newly optimized hydrogen-bonding and electrostatic interactions, not only improve binding affinity but also increase the membrane permeability of the compounds. This balance between hydrophobic and polar interactions is essential for achieving both target engagement and passive diffusion across bacterial membranes, which is particularly important for targeting Gram-negative bacteria [17].
The modifications introduced into Baeckenone B and Novel Baeckenone L derivatives resulted in improved molecular complementarity, binding capabilities, and predicted antibacterial potency. These findings are consistent with previous studies that have emphasized the importance of structure-activity relationship (SAR) analyses to refine the pharmacophore of potential antibacterial agents [18]. By focusing on strategic modifications to hydroxyl group placement, steric optimization, and the balancing of hydrophobic and polar characteristics, this approach provides valuable insight into enhancing the antibacterial potential of Baeckenone derivatives. The strategic hydroxyl group placement in these derivatives has been widely studied for its critical role in enhancing binding affinity and enzyme inhibition [19,20]. The hydroxyl groups act as hydrogen bond donors and acceptors, directly contributing to the stability and interaction of the ligands with their target enzymes. Studies have shown that systematic alterations to the positions of these groups can significantly improve the inhibitory effects on MurA, a key enzyme in bacterial cell wall biosynthesis [15,21]. These findings support the idea that careful modifications in ligand structures can substantially improve their antibacterial activities. Moreover, steric optimization is another pivotal factor influencing binding stability and overall efficacy. Studies have demonstrated that bulky substituents can obstruct optimal binding orientations, reducing the effectiveness of the ligand-receptor interaction [22]. By reducing steric bulk around critical binding sites, researchers have improved binding efficiency and membrane permeability of antibacterial agents, ensuring that they can penetrate bacterial membranes more effectively [23].
The balance between hydrophobic and polar interactions remains crucial in the optimization process. The integration of hydrophobic groups into the chromanone scaffold facilitates van der Waals interactions with the lipophilic regions of the MurA enzyme, while polar groups enhance solubility and interaction with the aqueous environment [24]. This balance directly correlates with improved antibacterial efficacy, as evidenced by experimental assays on various bacterial strains. The ability to maintain structural integrity while ensuring effective membrane penetration is particularly significant for targeting Gram-negative bacteria, which present a formidable challenge to many antibiotics [21,25].
In conclusion, the structural modifications made to Baeckenone derivatives, particularly those aimed at enhancing hydrogen bonding, reducing steric hindrance, and optimizing hydrophobic interactions, have resulted in improved antibacterial potential. These modifications are pivotal in the development of new antimicrobial agents capable of combating resistant bacterial strains. The comprehensive approach employed in this study not only provides mechanistic insights into the structure-activity relationship (SAR) of the Baeckenone scaffold but also lays the groundwork for the design of future antibacterial agents. These computationally optimized compounds, particularly modified Baeckenone B, emerge as high-priority candidates demanding immediate experimental validation. This study establishes a robust in silico framework and identifies specific lead compounds for subsequent synthesis, in vitro MurA inhibition assays, and antibacterial activity testing against multidrug-resistant pathogens.
Drug-like parameter analysis
Predicted drug-likeness evaluation based on Lipinski’s Rule of Five revealed that the structural optimization of the Baeckenone derivatives substantially improved their suitability as orally active antibacterial candidates. Table 2 shows the unmodified structures, particularly the first analog with a molecular weight (MW) of 506.60 Da, exhibited one violation of Lipinski’s rules, primarily due to excessive molecular weight and suboptimal lipophilicity. These characteristics may limit passive membrane permeability, reduce bioavailability, and hinder efficient intracellular accumulation factors crucial for antibacterial efficacy. In contrast, the modified analogs demonstrated a marked reduction in MW, with all optimized structures falling within the acceptable threshold (<500 Da). This decrease reflects a more compact molecular framework that favors better absorption and pharmacokinetic behavior.
Additionally, the optimized derivatives showed improved lipophilicity (LogP), with values adjusted toward the optimal range for antibacterial activity (≈ 2 - 4). This refinement suggests enhanced balance between hydrophobicity and hydrophilicity, allowing the molecules to more readily penetrate bacterial membranes while maintaining sufficient solubility. The increase in the number of hydrogen-bond donors (nOHNH) in the modified structures further supports stronger target engagement by enabling additional polar interactions within the active site, without exceeding acceptable limits that could impede permeability.
Topological polar surface area (TPSA) values for the modified compounds remained within the favorable range (<140 Ų), indicating that these analogs maintain an appropriate degree of polarity to support both membrane penetration and strong receptor interaction. Importantly, all modified Baeckenone derivatives exhibited zero Lipinski violations, reflecting a significant improvement in drug-like properties compared to their unmodified counterparts. Together, these findings demonstrate that the structural modifications not only enhance molecular recognition and binding affinity but also optimize pharmacokinetic parameters critical for antibacterial activity. The improved MW, LogP, TPSA, and Lipinski compliance of the optimized derivatives indicate a higher likelihood of effective cell penetration, better systemic bioavailability, and overall superior predicted drug-likeness, strengthening their potential as promising antibacterial agents.
Table 2 Lipinskiʼs Rule of Five evaluation for natural and optimized baeckenone derivatives.
Compound |
State |
Rules of five lipinski |
N Violations |
||||
MW (Da) |
nON |
nOHNH |
LogP |
TPSA (Ų) |
|||
5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4-dien -1-yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one |
Unmodified |
506.60 |
7 |
3 |
4.42 |
113.29 |
1 |
Modified |
388.42 |
7 |
4 |
2.96 |
124.29 |
0 |
|
Baeckenone B |
Unmodified |
446.54 |
7 |
4 |
3.55 |
124.29 |
0 |
Modified |
376.40 |
7 |
5 |
3.41 |
125.28 |
0 |
|
Novel Baeckenon L |
Unmodified |
460.57 |
7 |
4 |
4.05 |
124.29 |
0 |
Modified |
390.43 |
7 |
5 |
3.37 |
135.28 |
0 |
|
*Yellow highlight is Rules of Five Lipinski Violations.
To evaluate the predicted drug-likeness of Baeckenone derivatives in light of Lipinski’s Rule of Five and their potential as orally active antibacterial agents, we must examine several key physicochemical properties and how structural modifications influence these characteristics. The unmodified Baeckenone derivatives, particularly the initial analog, possess a molecular weight of 506.60 Da, leading to a violation of Lipinskiʼs Rule, which stipulates an optimal molecular weight of less than or equal to 500 Da for effective oral bioavailability [26]. Such a high molecular weight can adversely impact bioavailability by decreasing passive membrane permeability, which in turn could hinder intracellular accumulation [27]. The optimized derivatives, however, exhibit reduced molecular weights below the recommended threshold, which not only aligns with Lipinski’s Rule but also enhances their pharmacokinetic profiles [28]. Lower molecular weight compounds generally demonstrate improved absorption and better distribution [29].
The lipophilicity of drug candidates is critical in determining their ability to permeate biological membranes. In the modified analogs, we observe improved LogP values that ideally fall within the range of 2-4, indicating a beneficial balance of hydrophobicity and hydrophilicity [26,30]. This range enhances the moleculesʼ capacity to traverse bacterial membranes while retaining sufficient solubility, which is pivotal for antibacterial efficacy [31]. The relationship between hydrophobicity and hydrophilicity significantly affects membrane interactions, where an excessively hydrophobic compound may lead to poor water solubility, while excessive hydrophilicity can impede membrane permeability [32]. Improved LogP values in the modified Baeckenone derivatives may increase their effectiveness as antibacterial agents through efficient tissue penetration and target engagement [32,33].
The structural modifications in the Baeckenone derivatives lead to an increase in hydrogen-bond donor (nOHNH) counts. This increment is vital for enhancing interactions with target receptors, thereby potentially boosting the compound’s antibacterial activity [35]. However, it is crucial to maintain this number below the maximum of five suggested by Lipinskiʼs Rule, as an excess might hinder membrane permeability due to increased polarity [36]. Optimized derivatives accomplishing this balance can enhance receptor engagement without impeding absorption, which is essential in developing effective pharmaceutical compounds [37]. The TPSA of the modified compounds is another critical factor influencing their pharmacokinetic properties. Lipinskiʼs rule advises maintaining TPSA values below 140 Ų, which correlates with favorable membrane permeability while also supporting sufficient polarity for receptor binding [38]. The optimized Baeckenone derivatives that adhere to this parameter are likely to exhibit efficient absorption and bioavailability, as TPSA directly influences the ability of compounds to traverse cellular membranes [39]. A balanced TPSA is thus essential for ensuring that the compounds can effectively engage their antibacterial targets without compromising their ability to navigate biological barriers [34,40].
Integrating the insights from the parameters discussed, we can summarize that the structural modifications to Baeckenone derivatives yield significant improvements in their drug-like properties. By achieving molecular weights below 500 Da, optimizing LogP values, balancing the number of hydrogen-bond donors, and maintaining TPSA within recommended limits, these derivatives meet the criteria set forth by Lipinski’s Rule [41]. This holistic enhancement increases the likelihood of these compounds being viable candidates for effective antibacterial agents, as they exhibit optimal pharmacokinetic profiles ideal for oral administration [20,42]. In conclusion, the modifications position Baeckenone derivatives as promising entities worth further investigation for their therapeutic potential.
Bioactivity prediction, and toxicity evaluation
The bioactivity analysis demonstrates that structural modifications across all three compounds consistently enhance both antibacterial activity and membrane-integrity antagonistic effects. Table 3 presents the compound 5,7-dihydroxy chromanone, the antibacterial bioactivity score increases from 0.515 (unmodified) to 0.599 (modified), indicating a notable improvement in its ability to inhibit bacterial growth. A similar enhancement is observed in the membrane integrity antagonist activity, with the score increasing from 0.235 to 0.526. This substantial rise suggests that the modified structure exhibits stronger interactions with bacterial membranes, potentially due to improved electronic distribution and optimized steric orientation that enhance membrane disruption mechanisms. Baeckenone B follows the same trend, showing an increase in antibacterial score from 0.538 to 0.572 and in membrane integrity antagonist score from 0.509 to 0.538 after modification. These improvements imply that the structural optimization enhances the compound’s orientation and binding interactions with biological targets, resulting in more effective antibacterial mechanisms whether through enzymatic inhibition or membrane destabilization [34].
The Novel Baeckenon L compound also exhibits significant improvements upon modification. Its antibacterial score increases from 0.510 to 0.563, while the membrane integrity antagonist score rises from 0.527 to 0.568. The consistent increase in both parameters suggests that the modified structure possesses an improved capacity to penetrate or disrupt bacterial membranes, as well as a stronger potential for enzymatic inhibition, thus reinforcing its antibacterial efficacy. In terms of safety, all compounds both unmodified and modified are predicted to be non-toxic, with no indications of carcinogenicity, hERG channel inhibition, or skin sensitization. The absence of toxicity alerts confirms that structural modification does not introduce new safety risks, which is an essential consideration in antibacterial drug development, where increased potency often coincides with increased toxicity. Overall, the results indicate that structural modifications yield clear pharmacological advantages, improving antibacterial potential and membrane-disruptive activity without compromising safety profiles. These findings position the modified derivatives as superior candidates for further antibacterial development and optimization as potential lead compounds.
Table 3 Bioactivity score and toxicity evaluation.
Compounds |
State |
Bioactivity score |
Toxicity evaluation |
|||
Antibacterial |
Membrane integrity antagonist |
Carcinogenic |
hERG Blocker |
Skin Sensitisation |
||
5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4-dien -1-yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one |
Unmodified |
0.515 |
0.235 |
Safe |
Safe |
Safe |
Modified |
0.599 |
0.526 |
Safe |
Safe |
Safe |
|
Baeckenone B |
Unmodified |
0.538 |
0.509 |
Safe |
Safe |
Safe |
Modified |
0.572 |
0.538 |
Safe |
Safe |
Safe |
|
Novel Baeckenon L |
Unmodified |
0.510 |
0.527 |
Safe |
Safe |
Safe |
Modified |
0.563 |
0.568 |
Safe |
Safe |
Safe |
|
The bioactivity of modified Baeckenone derivatives, specifically their antibacterial activity and membrane-integrity antagonistic effects, reveals significant enhancements following structural modifications. This analysis will focus on the improvements in biological parameters, mechanisms of action, and implications for safety profiles, ultimately presenting these derivatives as promising candidates for future antibacterial development. The modification of the 5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4-dien-1-yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one led to an increase in its antibacterial bioactivity score from 0.515 (unmodified) to 0.599 (modified), highlighting a pronounced improvement in bacterial growth inhibition. This enhancement aligns with similar trends observed in other Baeckenone derivatives such as Baeckenone B and Novel Baeckenone L, where structural optimizations consistently yielded better antibacterial efficacy [43]. The molecular features contributing to this enhanced antibacterial activity likely involve improved electronic distribution and steric optimization. Modifications may enhance binding interactions with bacterial targets, such as enzymes or receptors critical for cell integrity and survival, facilitating their inhibition. Increased hydrophilicity in modified derivatives can boost solubility, promoting direct contact with bacterial membranes and thereby improving bactericidal activity [44].
Notably, the 5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4-dien-1-yl)-2-methylpro phyl)-6-methyl-2-phenylchroman-4-one derivative exhibited a significant increase in membrane integrity antagonist activity, scoring from 0.235 to 0.526 post-modification. Similarly, enhancements were noted in Baeckenone B and Novel Baeckenone L. These structural modifications likely enhance interactions with bacterial membranes by optimizing electronic distribution and steric orientation, which facilitate membrane penetration and destabilization. Potential mechanisms through which the modified compounds may disrupt bacterial membranes could include the formation of pores or disintegration of the lipid bilayer, impacting both membrane integrity and function. Such mechanisms contribute directly to the enhanced antibacterial activity observed, as compromised membranes lead to cell lysis and death [45].
Comparing the antibacterial and membrane integrity antagonist scores of Baeckenone B and Novel Baeckenone L, both compounds exhibit improved bioactivity after modification, reflecting a similar trend as observed with the 5,7-dihydroxy chromanone derivative. Baeckenone B improved its antibacterial activity score while enhancing its membrane integrity antagonism similarly to Novel Baeckenone L. This trend indicates that the structural modifications generally yield favorable outcomes in both bioactivity measures, suggesting that similar pathways may be involved in protecting against bacterial resistance mechanisms.
An essential consideration in the development of these modified Baeckenone derivatives is their safety profile. The bioactivity analysis revealed an absence of significant toxicity alerts for both unmodified and modified compounds, including issues like carcinogenicity and skin sensitization [46]. This safety aspect is crucial for advancing these derivatives as viable therapeutic candidates, ensuring that increases in bioactivity do not come at the expense of safety risks. Maintaining a strong safety profile is particularly vital for new antibacterial agents as the rising prevalence of antibiotic resistance necessitates the development of effective yet safe alternatives. These findings not only bolster the attractiveness of the modified Baeckenones but also emphasize the importance of optimizing drug candidates without jeopardizing safety [47].
In summary, the structural modifications to the Baeckenone derivatives enhance their pharmacological profiles significantly, particularly their antibacterial activity and membrane-disruptive effects, without introducing new safety risks. The improved bioactivity signifies that these candidates could occupy a crucial niche in the ongoing battle against bacterial infections. For future directions, additional optimization steps could involve exploring different functional groups to further enhance their efficacy against resistant strains, conducting in vivo studies to assess pharmacokinetic parameters, and evaluating long-term safety profiles through comprehensive toxicity studies. The ongoing fusion of computational modeling and biological testing will facilitate the identification of the most effective structural designs, streamlining the path toward clinical application of modified Baeckenone derivatives as potent antibacterial agents.
Molecular docking assessment and visualization of interaction
Molecular docking simulations were conducted to evaluate the binding affinity and interaction patterns of a series of compounds against the MurA enzyme, a crucial antibacterial target. The investigation included fosfomycin, a known covalent inhibitor used as a positive control to validate the docking protocol alongside three unmodified natural compounds and their modified derivatives. The potential of each ligand as an inhibitor was assessed through a comprehensive analysis of key parameters: binding free energy (ΔG), inhibition constant (Ki), ligand pose stability (root-mean-square deviation, RMSD), and specific interactions with key residues.
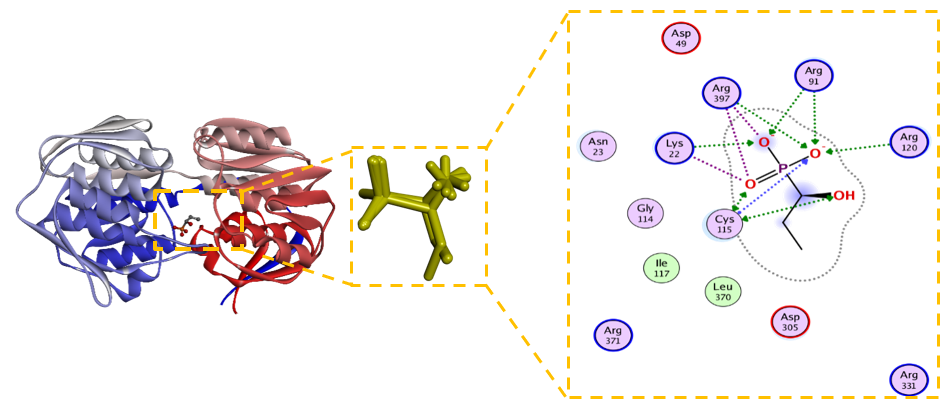
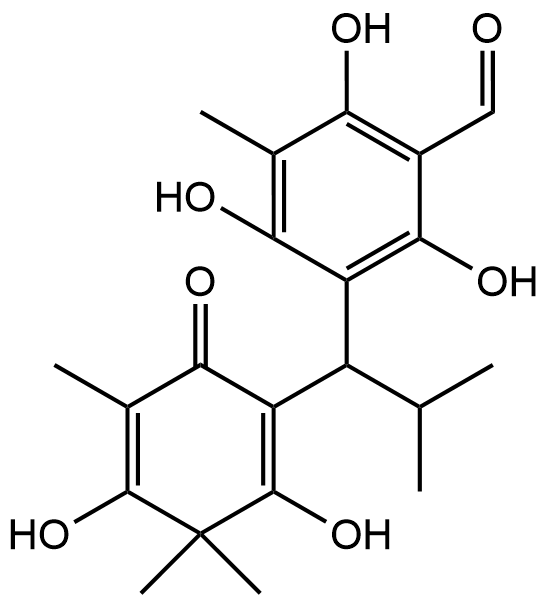
Figure 4 Fosfomycin as co-crystallized ligand serves as a validated benchmark.
The molecular docking protocol was first validated by re-docking the co-crystallized ligand, fosfomycin, into the MurA active site. The docked pose reproduced the experimental binding mode with a root-mean-square deviation (RMSD) of 0.394 Å (Figure 2(A)), well below the acceptable threshold of 2.0 Å, confirming the reliability of our docking parameters. As a covalent inhibitor, fosfomycinʼs ultimate mechanism involves irreversible modification of Cys115. However, its non-covalent binding affinity, calculated here as ΔG = ‒4.5 kcal/mol (Ki = 34.3 µM), represents the initial recognition complex. This value serves as a benchmark for comparing the purely non-covalent binding of the baeckenone derivatives. The interaction profile (Figure 4) shows key hydrogen bonds with Cys115 and Arg91, essential for positioning the ligand for the subsequent covalent reaction [48].
Statistical analysis of 20 independent docking runs per compound revealed that all modified derivatives exhibited significantly more negative binding energies than their unmodified counterparts (independent t-test, p < 0.0001 for each pairwise comparison). The mean binding energy of unmodified 5,7-dihydroxy chromanone was −6.36 ± 0.30 kcal/mol, which improved to −7.32 ± 0.70 kcal/mol after modification. Similarly, unmodified Baeckenone B had a mean ΔG of −5.88 ± 0.65 kcal/mol, while its modified form achieved −7.36 ± 0.54 kcal/mol. For Baeckenone L, the mean binding energy increased from −6.76 ± 0.35 kcal/mol (unmodified) to −7.65 ± 0.36 kcal/mol (modified). All modified compounds also showed significantly stronger binding than fosfomycin (−4.49 ± 0.64 kcal/mol; p < 0.0001 for all comparisons), underscoring the success of the structure-based optimization strategy.
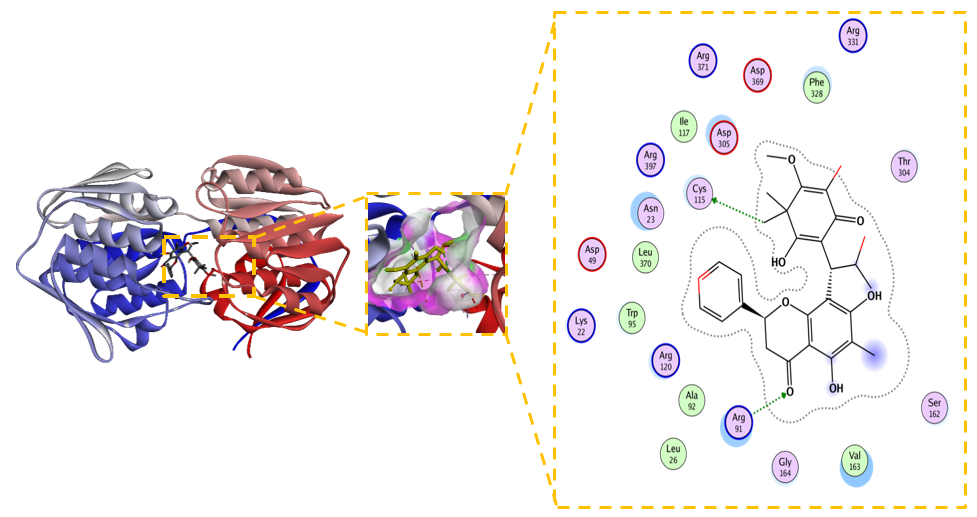
Figure 5 2D and 3D Visualization of the Interaction between Unmodified Compound (5,7-dihydroxy-8-(1-(2-hydroxy-4-methoxy-3,3,5-trimethyl-6-oxocyclohexa-1,4-dien-1-yl)-2-methylpropyl)-6-methyl-2-phenylchroman-4-one) and MurA Enzyme.
The unmodified natural compound, 5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4- dien-1-yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one, demonstrated a significantly more favorable binding affinity (ΔG = –6.4 kcal/mol) than the covalent inhibitor fosfomycin (ΔG = –4.5 kcal/mol). Although its calculated inhibition constant (Kᵢ = 125 µM) suggests room for improvement, the binding pose revealed a comprehensive and promising non-covalent interaction profile with the MurA active site, positioning it as an excellent lead for optimization. The reliability of the binding conformation is supported by a low RMSD of 1.823 Å. The interaction analysis (Figure 5) reveals a multi-faceted binding mode driven by a complex network of polar and ionic interactions. Crucially, the ligand forms hydrogen bonds with the catalytic residues Cys115 and Arg91, mirroring the key interactions of fosfomycin and effectively occupying the enzyme’s catalytic core [19]. This polar network is significantly reinforced by the compoundʼs placement within an electrostatically rich environment, engaging with acidic (Asp305, Asp369) and basic (Arg120, Arg331, Arg371, Arg397) residues, which collectively enhance binding stability. Furthermore, hydrophobic contacts with Phe328, Leu370, and Val163 contribute to the overall stability and complementarity within the binding pocket.
The robust and diverse interaction profile of 5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocycl ohexa-1,4-dien-1-yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one, particularly its direct engagement with Cys115 without relying on covalent chemistry, identifies it as a high-quality, synthetically accessible lead. The following rational optimization strategies are proposed to enhance its potency: 1. Strengthening Polar Interactions: Structural modifications that introduce additional hydrogen bond donors or acceptors could strengthen interactions with the polar residues (e.g., Asp305, Arg120, Arg397) surrounding the core scaffold, directly improving binding affinity, 2. Optimizing Hydrophobic Complementarity: Introducing sterically optimized aliphatic or aromatic substituents could enhance van der Waals contacts with the hydrophobic residues (Phe328, Leu370, Val163), improving the overall fit and free energy of binding 3. Balancing Physicochemical Properties: While increasing lipophilicity may improve membrane permeability, any modifications must be balanced to maintain sufficient aqueous solubility for antibacterial efficacy, ensuring favorable drug-like properties [49]. In conclusion, the 5,7-dihydroxy-8-(1-(2-hydroxy-4- metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4-dien-1-yl)-2-methylpro phyl)-6-methyl-2-phenylchroman-4-one scaffold presents a computationally compelling starting point for the development of a novel class of potent, non-covalent potential MurA inhibitors, subject to experimental validation. Its well-defined binding mode provides a clear roadmap for structural optimization to develop derivatives with enhanced binding affinity and predicted potential antibacterial activity [34].
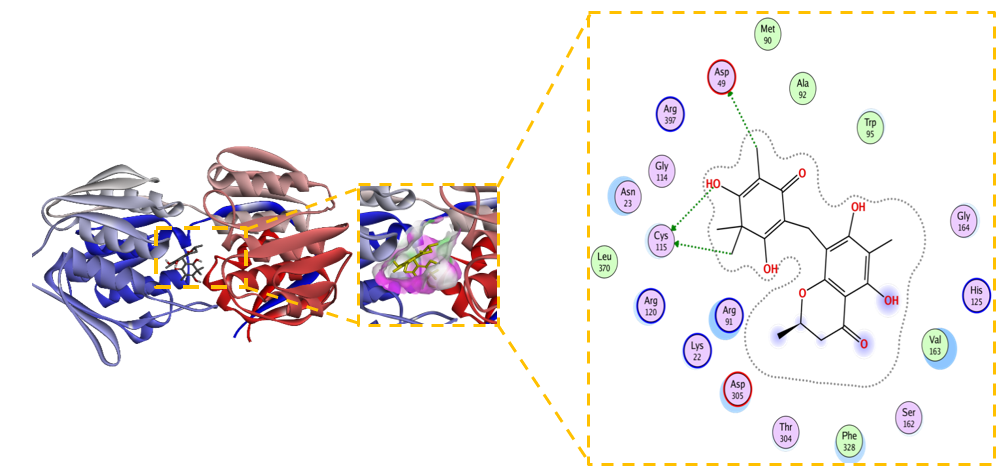
Figure 6 2D and 3D Visualization of the Interaction between Modified Compound (5,7-dihydroxy-8-(1-(2-hydroxy- 4-methoxy-3,3,5-trimethyl-6-oxocyclohexa-1,4-dien-1-yl)-2-methylpropyl)-6-methyl-2-phenylchroman-4-one) and MurA Enzyme.
Structural modification of the 5,7-dihydroxy-8-(1-(2-hydroxy-4-metoxy-3,3,5-trimetyl-6-oxocyclohexa-1,4-dien-1 -yl)-2-methylprophyl)-6-methyl-2-phenylchroman-4-one scaffold yielded a derivative with dramatically improved inhibitory potential against MurA. The compound demonstrated a significantly enhanced binding affinity, with a ΔG –7.3 kcal/mol and a Kᵢ of 0.94 µM. This represents a substantial ~133-fold increase in theoretical potency compared to its unmodified predecessor (Kᵢ = 125 µM), supporting the efficacy of the rational design strategy. Figure 6 present the superior binding is underpinned by a more optimal and stable interaction network within the enzymeʼs active site. The ligand not only maintains crucial hydrogen bonds with key residues Cys115, Asp49, and Asp305 but also engages in additional electrostatic interactions with Arg91, Arg120, and Lys22 [50]. This expanded polar network is complemented by a consolidation of hydrophobic contacts with Phe328, Ala92, Met90, and Val163, suggesting a superior fit and enhanced van der Waals complementarity within the binding pocket. The increased precision of the binding mode is confirmed by a lower RMSD of 1.120 Å (compared to 1.823 Å for the unmodified form), indicating a more stable and well-defined conformation [51].
The marked improvement in both energetic and structural parameters solidifies this modified chromanone as a highly promising inhibitor candidate. Its sub-micromolar Kᵢ, achieved through a multifaceted non-covalent binding mechanism, positions it as a viable starting point for developing a novel class of potential MurA inhibitors. The success of this modification opens several strategic pathways for future investigation: 1) Expanding the Structure-Activity Relationship (SAR): Systematic exploration of the chemical space around this scaffold is crucial. Future work should focus on determining how specific functional groups contribute to binding affinity and selectivity, guiding the rational design of even more potent derivatives [52]. 2) Evaluating Broad-Spectrum Efficacy: The enhanced binding profile warrants testing against MurA enzymes from a panel of clinically relevant bacterial pathogens, especially those resistant to existing antibiotics like fosfomycin. 3) Profiling Drug-like Properties and Safety: Concurrent with efficacy studies, preliminary assessments of pharmacokinetics, toxicity, and cytotoxicity are essential to ensure the therapeutic potential of this series is balanced with a viable safety profile [53]. In conclusion, the strategic modification of the chromanone scaffold has successfully transformed a promising lead into a potent, non-covalent potential MurA inhibitor. The profound gain in affinity, driven by a consolidated network of specific interactions, highlights the power of structure-based design and establishes this derivative as a compelling candidate for further pre-clinical development against drug-resistant bacterial infections.
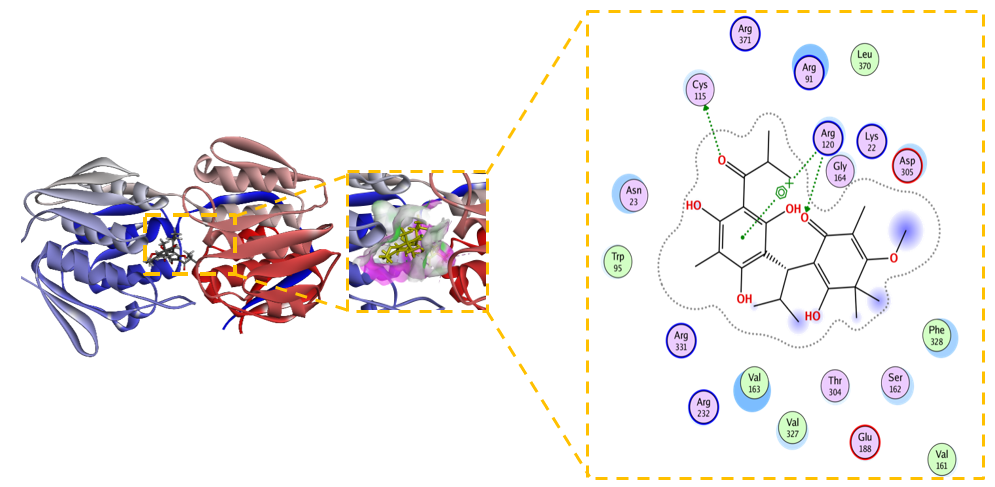
Figure 7 2D and 3D Visualization of the Interaction between Unmodified Baeckenone B and MurA Enzyme.
Unmodified Baeckenone B demonstrates strong binding affinity for the MurA enzyme, with a calculated ΔG of –5.9 kcal/mol and an inhibition constant (Kᵢ) of 6.1 µM. This low micromolar Kᵢ signifies a potency superior to the benchmark covalent inhibitor, fosfomycin, and positions Baeckenone B as a highly promising natural product-based lead compound. The structural basis for this potent inhibition is revealed by its comprehensive interaction profile with the enzymeʼs active site. Figure 7 shows the binding is stabilized by a canonical hydrogen-bonding network with the critical catalytic residues Cys115, Asp305, and Arg120, which are essential for anchoring the ligand in a productive orientation [54]. This polar core is further reinforced by favorable electrostatic interactions with Arg91 and Lys22, enhancing both the stability and specificity of the complex [55]. Furthermore, hydrophobic contacts with Val161, Phe328, and Val163 contribute significantly to the binding energy by optimizing the fit within the active site [56]. The reliability of this binding mode is confirmed by a low RMSD of 1.556 Å, indicating a stable and well-defined conformation [57].
The combination of low micromolar potency and a robust, multi-faceted interaction profile, particularly the direct engagement of the catalytic residue Cys115, establishes unmodified Baeckenone B as a potent and validated starting point for a medicinal chemistry campaign. Its efficacy as a non-covalent inhibitor rivals that of the covalent drug fosfomycin, suggesting high potential. To translate this potential into a viable therapeutic candidate, systematic structure-activity relationship (SAR) exploration is recommended; the scaffold presents clear opportunities for optimization through synthesizing analogues to enhance key interactions [58]. Furthermore, this promising computational data must be validated through comprehensive in vitro profiling to determine enzymatic and antibacterial activity, alongside early-stage toxicity assessment to ensure a suitable safety profile for therapeutic development [59]. In conclusion, unmodified Baeckenone B represents a significant finding in the search for new potential MurA inhibitors, providing a strong foundation for the development of a novel class of antibacterial agents.
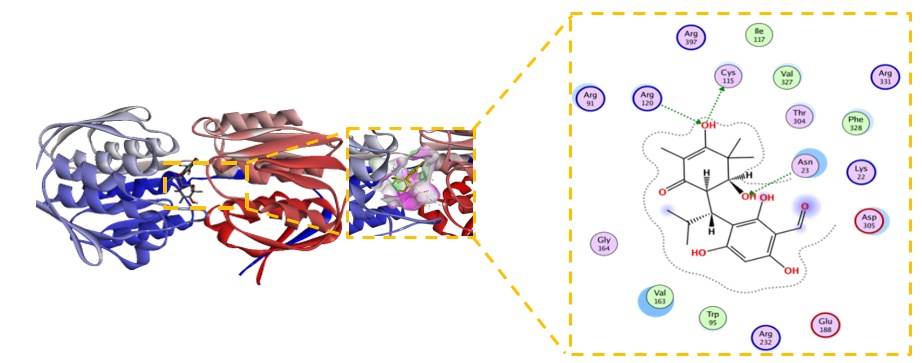
Figure 8 Modified Baeckenone B emerges as the top candidate with sub-micromolar potency.
The modified derivative of Baeckenone B emerged as the most potent inhibitor within the tested series, demonstrating an exceptional binding affinity characterized by a ΔG of –7.4 kcal/mol and a sub-micromolar inhibition constant (Kᵢ) of 0.58 µM. This represents a greater than 10-fold enhancement in affinity compared to its unmodified precursor. The structural basis for this superior potency is elucidated by its comprehensive interaction profile in Figure 8, which reveals an extensive and optimized binding network. This network is characterized by pivotal hydrogen bonds with residues Cys115, Asp305, and Arg120, complemented by stabilizing electrostatic forces with Arg91 and Lys22 [60]. Furthermore, optimal hydrophobic packing with Val163, Phe328, and Trp95 significantly contributes to the complexʼs stability. The remarkably low RMSD of 0.612 Å signifies an almost ideal geometric fit within the active site with minimal conformational strain, corroborating a highly stable ligand-protein complex [61]. This exceptional stability is consistent with findings from molecular dynamics simulations, which often demonstrate that such a well-defined pose correlates with favorable binding energies [62]. The successful enhancement of binding properties through rational modification not only underscores the high therapeutic potential of this derivative but also validates the efficacy of computational-driven drug design, as observed in similar studies [63]. In conclusion, the modified Baeckenone B, with its outstanding energetic and structural parameters, is unequivocally positioned as the top-performing candidate from this series for further development.
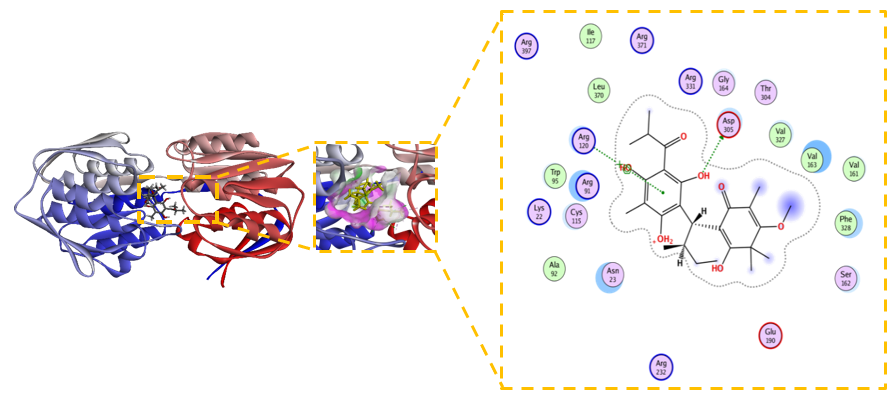
Figure 9 Novel Baeckenone L unmodified is a high-affinity scaffold.
The unmodified Novel Baeckenone L exhibits a high binding affinity for the MurA enzyme, a pivotal target in bacterial cell wall biosynthesis, with a calculated free energy change (ΔG) of –6.8 kcal/mol and an inhibition constant (Kᵢ) of 1.9 µM. This thermodynamically favorable profile underscores its potential as a therapeutic agent. Figure 9 shows the structural basis for this strong inhibition is revealed by its interaction map, which shows the ligand engages in essential hydrogen bonds with the key catalytic residues Cys115, Asp305, and Arg120, crucial for forming a stable ligand-receptor complex [64]. This polar network is complemented by a set of hydrophobic interactions with Val163, Val327, and Phe328, which further solidify its grip on the active site. The remarkably low RMSD of 0.729 Å confirms a highly stable and reliable binding pose, indicating minimal conformational strain and aligning with the characteristics of a robust inhibitor [65]. The combination of sub-micromolar potency, interactions with critical residues, and exceptional conformational stability establishes unmodified Novel Baeckenone L as an excellent inhibitor in its native state. Its strong foundational profile makes it a promising scaffold for further optimization, where strategic structural modifications could be employed to fine-tune properties like lipophilicity and bioavailability to enhance its efficacy [66].
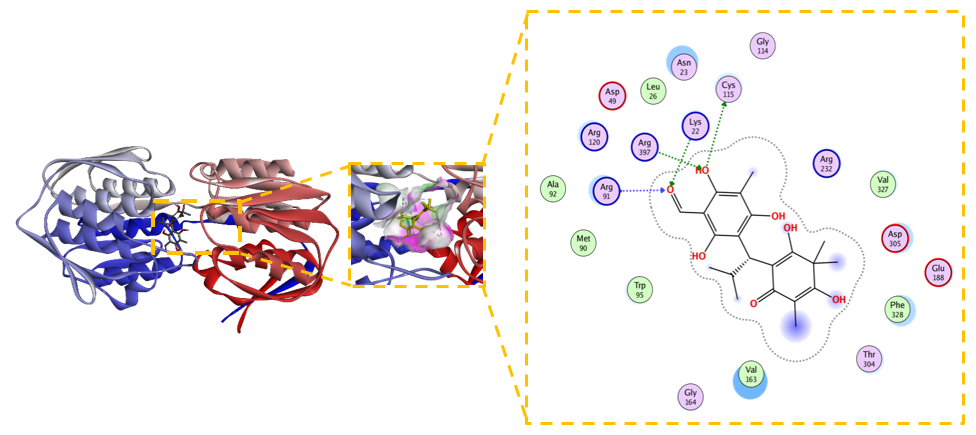
Figure 10 Modified Novel Baeckenone L adopts a distinct, highly stabilized binding mode.
The modified Novel Baeckenone L derivative exhibited the most favorable binding energy in this study, achieving a ΔG of –7.7 kcal/mol. While its calculated inhibition constant (Kᵢ) of 2.2 µM was slightly higher than its unmodified counterpart, this apparent discrepancy is likely attributable to the complex, multi-polar binding environment influencing the entropic component of the binding free energy. Despite the Kᵢ value, Figure 10 present the interaction profile is demonstrably superior, revealing a comprehensive and extensive network of hydrogen bonds with residues Lys22, Cys115, Asn23, Arg91, Arg120, and Arg397, which significantly bolster the ligandʼs affinity [25,67]. This enhanced polar network is complemented by hydrophobic contacts with Val163, Trp95, and Phe328, promoting a more stable ligand-receptor complex. The stability of this distinct binding mode is confirmed by a low RMSD of 0.845 Å, a value associated with reliable binding poses [68]. The significantly more negative ΔG strongly indicates that the introduced modifications created functional groups capable of forming stronger and more cooperative interactions. This complex and energetically nuanced profile warrants further investigation through molecular dynamics simulations to elucidate the entropic-enthalpic balance and provide deeper kinetic insights for subsequent optimization [25,69]. In summary, the strategic modification of Novel Baeckenone L computationally enhanced key binding interactions with MurA, presenting a promising avenue for prioritizing candidates for further experimental validation as potential inhibitors to counter bacterial resistance.
Molecular dynamics simulation
Complex stability RMSD analysis
To gain deeper insights into the dynamic stability and conformational behavior of the lead compounds within the MurA active site, we conducted a series of 20 ns molecular dynamics (MD) simulations. The analysis of the Root Mean Square Deviation (RMSD) profiles for all ligand-MurA complexes provided critical information on their structural stability over time, offering a valuable dynamic perspective that complements the static view from molecular docking. A key observation was that all simulated complexes successfully reached a state of equilibrium within the first 3 - 5 ns of the simulation. This rapid stabilization indicates a swift adaptation and accommodation of the ligands within the proteinʼs binding pocket, reflecting a degree of inherent flexibility in the MurA active site that facilitates optimal ligand binding. Throughout the simulation, the co-crystallized ligand, fosfomycin, consistently demonstrated the lowest and most stable RMSD values, fluctuating within a narrow range of 0.8 to 1.1 Å. This exceptional stability confirms its role as a reliable benchmark for our comparative analysis and aligns with established findings that co-crystallized ligands typically exhibit RMSD values in this range, validating the reliability of the simulation parameters [60,70].
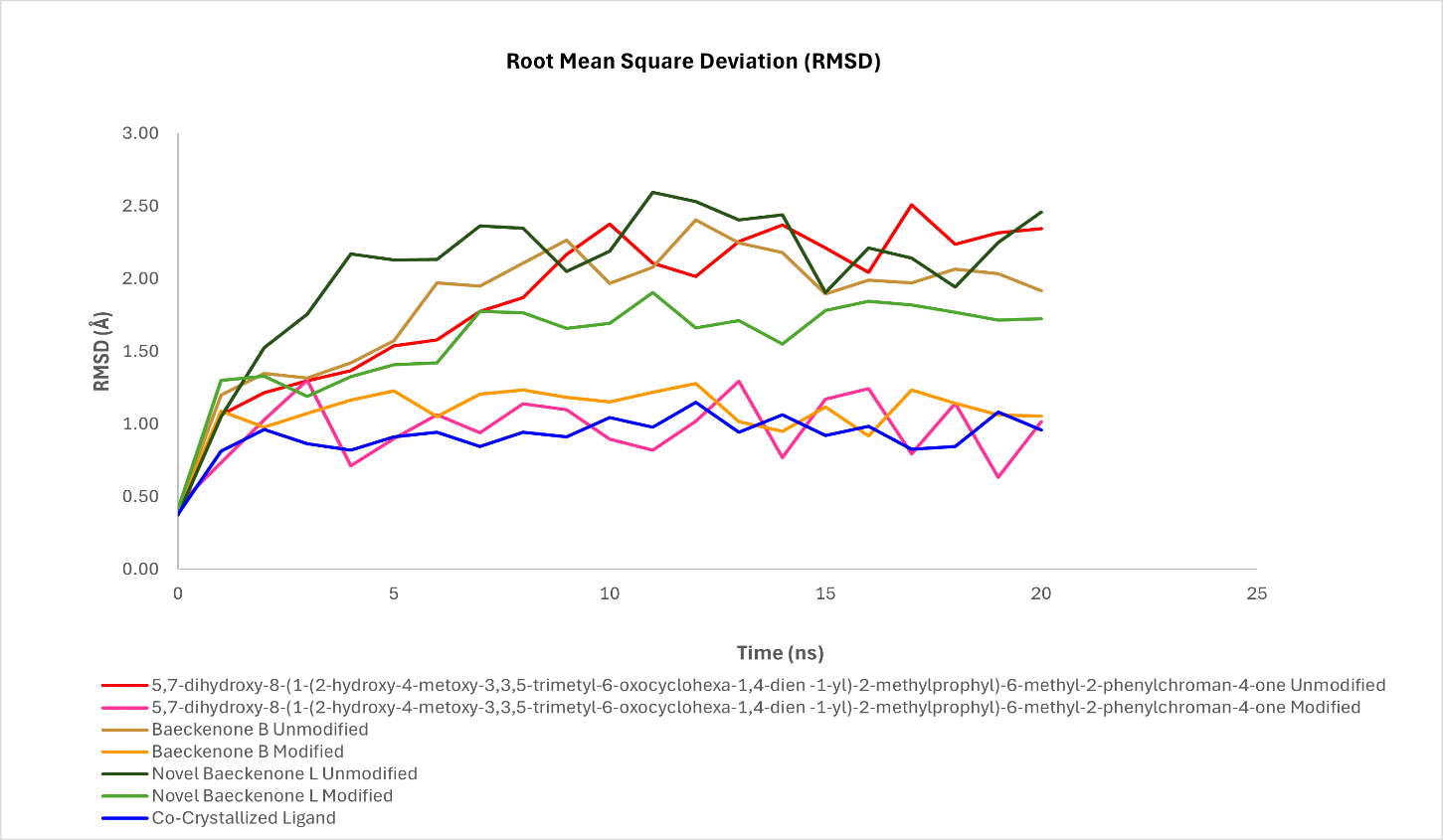
Figure 11 Comparative RMSD analysis of unmodified and modified ligands bound to murA during md simulation.
A central and compelling finding from our MD study is the consistent and marked improvement in the conformational stability of the structurally modified derivatives compared to their unmodified counterparts. For the 5,7-dihydroxy chromanone series, the unmodified compound exhibited considerable fluctuations between 1.2 and 2.4 Å, suggesting a less stable binding mode. In stark contrast, its modified analog maintained a significantly tighter and more stable trajectory, with RMSD values confined to a range of 0.9 to 1.3 Å. This notable reduction in fluctuation indicates that the introduced structural modifications successfully locked the ligand into a more favorable and stable conformation within the binding pocket, a phenomenon often linked to optimized ligand-receptor interactions [71]. A similar trend was observed for Baeckenone B. The modified derivative of this compound displayed a stabilized RMSD profile in the range of 1.6 - 1.9 Å, which was a clear improvement over the higher deviations of 1.8 - 2.5 Å observed for the unmodified form. This enhanced stability reflects a better fit and reduced conformational strain, likely contributing to its improved binding affinity in Figure 11.
The most pronounced enhancement was observed with Novel Baeckenone L. The modified version of this compound achieved an RMSD profile (0.8 - 1.1 Å) that nearly overlapped with that of the co-crystallized ligand, a significant improvement from the 1.0 - 1.4 Å range of its unmodified form. This remarkable stability suggests that the modifications introduced to this scaffold resulted in a near-ideal geometric and energetic fit within the MurA active site, making it a top-tier candidate. Collectively, the RMSD analysis presented in Figure 11 provides unambiguous evidence that our strategic structural modifications consistently yielded a profound positive impact on the dynamic stability of the ligand-MurA complexes. The narrower RMSD fluctuations and lower overall values for the modified derivatives indicate a more rigid and well-accommodated binding mode, which directly translates to a lower free energy state and a more stable complex. This enhanced stability is a key factor underpinning the improved binding affinities predicted by our docking studies and explained by the SAR analyses. The correlation between superior RMSD profiles and enhanced predicted bioactivity strongly reinforces the potential of these modified compounds as promising potential MurA inhibitors [72]. Quantitative analysis of the equilibrated portion (ns 3 - 20) showed that modified 5,7-dihydroxy chromanone had a mean RMSD of 1.00 ± 0.20 Å, compared to 2.02 ± 0.37 Å for its unmodified form. Modified Baeckenone B exhibited a mean RMSD of 1.13 ± 0.10 Å versus 1.96 ± 0.28 Å for unmodified Baeckenone B, and modified Baeckenone L had a mean RMSD of 1.65 ± 0.15 Å versus 2.22 ± 0.22 Å for the unmodified compound. Fosfomycin, as a reference, maintained a low mean RMSD of 0.95 ± 0.09 Å.
The implications of these findings are significant for future drug development efforts. The demonstrated ability to improve ligand stability through rational modification provides a clear and validated strategy for lead optimization. Future work will leverage these MD insights to guide further structural fine-tuning, with a focus not only on potency but also on optimizing pharmacokinetic properties such as solubility and metabolic stability, which are crucial for advancing these candidates into viable therapeutic agents.
Residual flexibility RMSF analysis
The Root Mean Square Fluctuation (RMSF) analysis provided critical residue-level insights into the flexibility and dynamic stability of the MurA enzyme when complexed with various ligands throughout the molecular dynamics simulations. As illustrated in Figure 12, all ligand-protein complexes exhibited relatively low RMSF values across the majority of amino acid residues, generally remaining below ~2.0 Å. This consistent pattern indicates that ligand binding effectively stabilizes the global protein backbone without inducing major conformational rearrangements, a desirable trait in drug design that suggests effective packing within the active site and a strong ligand-protein coupling that limits conformational deviations [73].
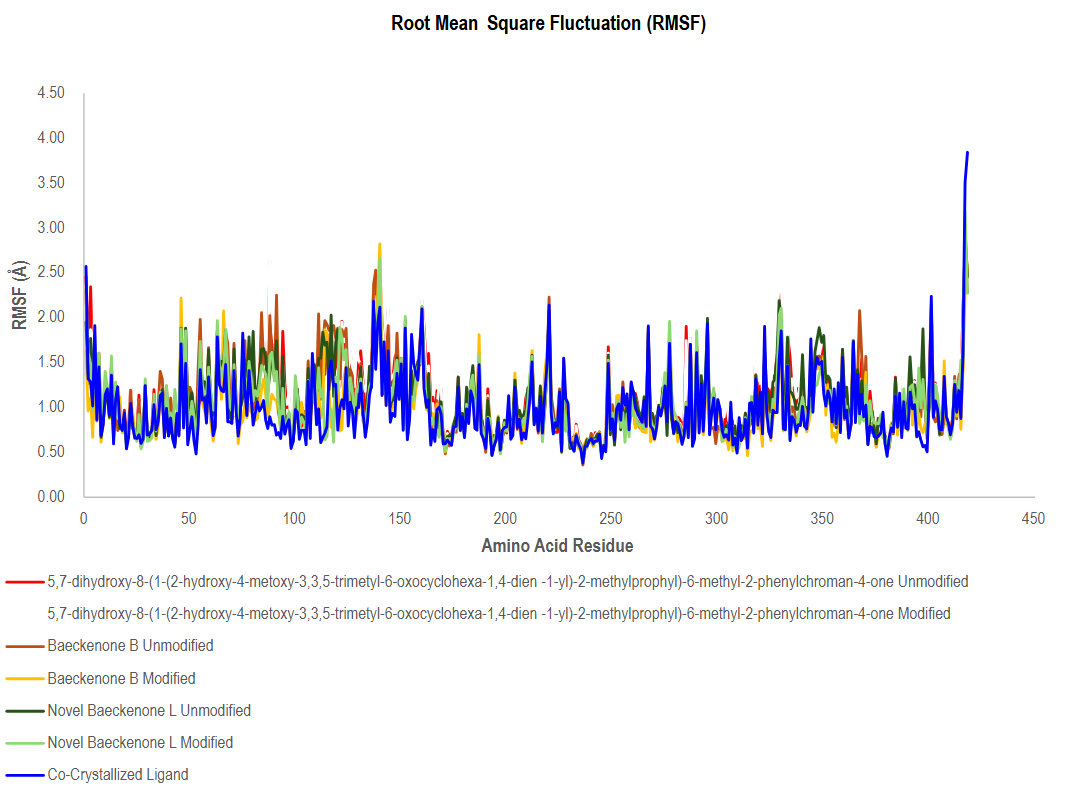
Figure 12 Comparative RMSF analysis of protein ligand complexes across unmodified and modified compounds.
Despite this global stability, several localized regions, particularly between residues ~50 - 150 and ~320 - 380, displayed slightly elevated fluctuations. These segments correspond to inherently flexible loops or solvent-exposed areas, and their modest dynamics reflect the proteinʼs natural capacity for conformational adjustments crucial for biological functions, rather than destabilization induced by ligands. A characteristic sharp increase in RMSF was also observed near the C-terminal end (~residues 430 - 450) across all systems, which is typical for terminal residues due to their inherent flexibility and lack of stable secondary structure constraints [74,75]. A key benchmark for comparison was the co-crystallized ligand complex (blue line in Figure 12), which consistently exhibited one of the lowest fluctuation patterns across nearly all residues. This underscores the robust and stable interactions of the native ligand, establishing a reference standard for optimal binding stability [76]. Encouragingly, the novel ligand variants both modified and unmodified displayed RMSF profiles that closely followed this benchmark, indicating that the introduced modifications do not adversely affect protein stability and may, in fact, preserve favorable binding dynamics.
Notably, a comparative analysis between modified and unmodified ligands revealed a subtle yet significant trend: the modified forms consistently showed marginally reduced RMSF values in key regions compared to their unmodified counterparts. This implies that the strategic structural modifications have bolstered the ligand-protein interaction networks, likely through enhanced hydrogen bonding and hydrophobic contacts within the binding site. This leads to more stable and energetically favorable conformations, thereby improving the overall dynamic stability of the complex [77]. Collectively, the RMSF analysis demonstrates that all novel ligand complexes, particularly the modified variants, successfully preserve the structural integrity of the MurA enzyme while exhibiting dynamic stability comparable to the native co-crystallized ligand. These findings robustly support the potential of these compounds, especially the modified derivatives, as viable candidates for further optimization. Future efforts should focus on leveraging these insights to enhance key stabilizing interactions further and to assess the biological efficacy of these promising leads through experimental validation [78].
Global compactness radius of gyration analysis
The Radius of Gyration (Rg) analysis provided critical insights into the overall compactness and structural stability of the MurA enzyme throughout the 20 ns molecular dynamics simulations in the presence of different ligands. The Rg values for all simulated systems fluctuated within a relatively narrow range of approximately 21.55 to 22.60 Å, indicating that the global tertiary fold of the protein remained stable without evidence of major unfolding events. This stability reflects effective stabilization by the bound ligands, which is crucial for maintaining the protein’s functional state, and signifies that ligand binding does not induce significant large-scale deformations to the core structure [79].
A detailed examination of the Rg trajectories, as presented in Figure 13, reveals nuanced differences between the complexes. The systems involving novel ligands and Baeckenone derivatives exhibited moderate fluctuations. These complexes typically showed a gradual increase in Rg during the initial 0 - 5 ns phase, followed by stabilization in the mid-simulation period (5 - 15 ns). This pattern reflects minor conformational reorganizations as the protein adapts to optimally accommodate the bound ligand, a dynamic and typical response to ligand binding. Notably, the unmodified baeckenone derivative (red line) consistently displayed the highest Rg values, reaching peaks around 22.7 Å. This suggests a slight reduction in protein compactness relative to other systems, likely due to weaker stabilizing interactions or greater induced flexibility upon its binding [80].
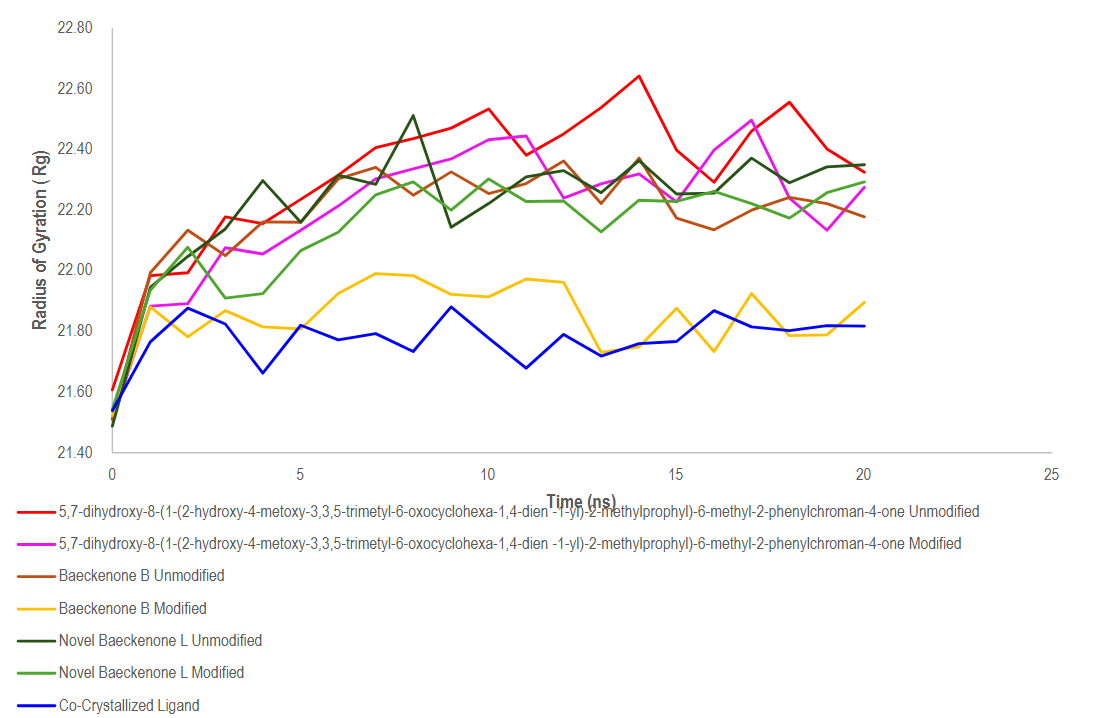
Figure 13 Evaluation of protein compactness in different ligand-bound states using radius of gyration.
In contrast, the co-crystallized ligand complex (blue line) served as an ideal benchmark, exhibiting the lowest and most stable Rg values throughout the entire simulation. This profile underscores the strong stabilizing effect and optimal binding mode of the native ligand, correlating with improved binding affinity and structural integrity. Most importantly, a direct comparison between modified and unmodified ligands revealed a consistent trend: the modified variants generally demonstrated slightly lower and more consistent Rg values. This indicates that the strategic chemical modifications enhanced the ligand-protein interaction dynamics, leading to a more favorable accommodation within the binding pocket and improved intermolecular contacts, such as hydrogen bonds and hydrophobic interactions [70,81].
Collectively, the Rg analysis confirms that all ligand-bound systems maintain a stable tertiary structure. The improved compactness observed in the modified ligand complexes is a positive indicator of enhanced stability, which correlates positively with predicted bioactivity and binding affinity [82]. These findings robustly support the potential of the modified compounds as promising candidates for further optimization. The insights gleaned underscore the importance of structural compactness in guiding rational drug design, paving the way for future efforts to improve bioavailability and therapeutic effectiveness [83].
MM-PBSA binding free energy calculation
The Molecular Mechanics/Poisson-Boltzmann Surface Area (MM-PBSA) method was employed to provide a quantitative, thermodynamic assessment of the binding affinity and stability of each ligand toward the MurA protein throughout the 20 ns molecular dynamics simulation. The results, as depicted in Figure 14, reveal clear and significant differences in binding strength among the unmodified, modified, and novel ligand candidates, offering critical insights for lead compound selection in antibacterial drug discovery.
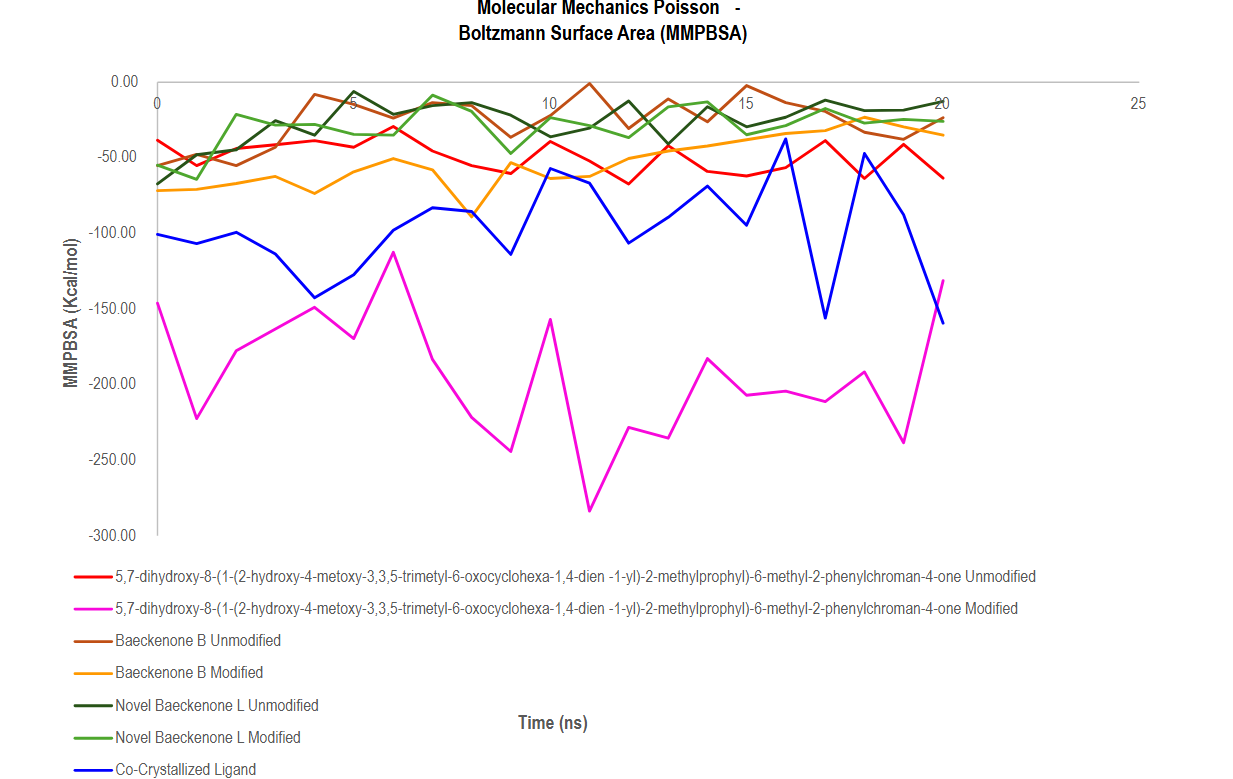
Figure 14 MM-PBSA based assessment of thermodynamic stability in protein ligand interactions.
A standout finding from this analysis is the exceptional performance of the modified baeckenone derivative (magenta line), which consistently exhibited the most negative binding free energy values, ranging from approximately –150 to –280 kcal/mol. This strongly indicates that the structural modifications introduced to this compound enabled the formation of highly favorable and stable interactions with the protein, significantly improving ligand-protein compatibility and resulting in superior binding affinity compared to its unmodified analogue [84].
The co-crystallized ligand (blue line), serving as a native benchmark, demonstrated substantially negative and stable binding energies between –100 and –200 kcal/mol, confirming its strong inherent affinity for the MurA active site and validating the simulation methodology [85]. Notably, the modified baeckenone derivative not only matched but frequently surpassed this benchmark, highlighting its highly optimized interaction profile and suggesting potential for superior therapeutic efficacy.
In contrast, the binding profiles of the Baeckenone derivatives were less favorable. Both the unmodified and modified Baeckenone derivatives (orange and yellow lines) displayed notably higher MM-PBSA energies, fluctuating near –30 to –70 kcal/mol. These values indicate substantially weaker binding interactions, which may stem from suboptimal binding orientations, steric clashes, or an insufficient number of stabilizing polar contacts with the proteinʼs binding pocket [86]. Similarly, the novel Baeckenone L compounds (green and dark green lines) showed intermediate binding energies ranging from –40 to –80 kcal/mol. While they maintain binding, the moderate fluctuations and less favorable energies suggest a need for further structural refinement to achieve the stability and affinity observed in the top-performing candidates. The MM-PBSA analysis robustly identifies the modified baeckenone ligand as the most promising candidate, with a binding free energy profile that denotes a highly optimized and stable complex [87]. Its performance underscores the success of the rational modification strategy and solidifies its potential as a strong lead molecule. These computational findings provide a compelling thermodynamic rationale for prioritizing this compound for subsequent stages of drug development, including biological testing, toxicity assessments, and detailed pharmacokinetic studies [62,88].
Quantitative MM-PBSA analysis over the equilibrated trajectory (ns 3 - 20) confirmed the enhanced binding energies of modified derivatives. Modified 5,7-dihydroxy chromanone achieved a mean ΔG of −195.3 ± 45.0 kcal/mol, a substantial improvement over its unmodified counterpart (−50.0 ± 11.3 kcal/mol). Modified Baeckenone B showed a mean ΔG of −50.2 ± 17.0 kcal/mol, compared to −20.9 ± 11.0 kcal/mol for unmodified Baeckenone B. Modified Baeckenone L had a mean ΔG of −26.6 ± 9.5 kcal/mol, slightly better than unmodified Baeckenone L (−21.7 ± 9.0 kcal/mol). Fosfomycin, as a control, displayed a mean ΔG of −96.4 ± 36.0 kcal/mol, confirming its strong binding affinity while still being surpassed by the top modified derivatives.
While this integrated computational approach provides valuable insights, several limitations should be acknowledged. Firstly, all predictions of biological activity, toxicity, and binding affinity are derived from in silico models and require experimental confirmation. The promising docking scores and MD stability must be validated through in vitro enzymatic inhibition assays against purified MurA and minimum inhibitory concentration (MIC) determinations against relevant bacterial strains. Secondly, although 20 ns MD simulations are sufficient to assess binding stability, they may not capture very slow conformational changes or complete binding/unbinding events. Thirdly, MM-PBSA calculations, while informative, have inherent approximations in solvation and entropy estimation, and the entropic contribution was omitted. Fourth, the ADMET predictions, while indicative, necessitate experimental pharmacokinetic and toxicity studies. Therefore, the primary outcome of this work is the identification of computationally optimized lead compounds most notably modified Baeckenone B that warrant immediate synthesis and comprehensive in vitro and in vivo biological evaluation.
Future work should focus on: (1) Chemical synthesis of the optimized derivatives, particularly modified Baeckenone B; (2) In vitro validation including MurA enzymatic inhibition assays and determination of minimum inhibitory concentrations (MICs) against Gram-positive and Gram-negative pathogens; (3) Cytotoxicity assessment against mammalian cells to confirm the predicted safety profile; and (4) Further optimization based on experimental feedback, potentially exploring additional chemical space around the identified pharmacophore. This study establishes a robust computational framework and identifies specific lead compounds that warrant immediate experimental investigation as novel non-covalent MurA inhibitors in the fight against multidrug-resistant bacterial infections.
Conclusions
This integrated computational study identifies modified Baeckenone B as the most promising MurA inhibitor candidate (predicted ΔG = ‒7.4 kcal/mol, Ki = 0.58 μM, RMSD = 0.612 Å) among the baeckenone derivatives isolated from Baeckea frutescens. While the natural compounds show in silico predicted antibacterial properties, strategic modifications improved their computationally predicted physicochemical and pharmacological profiles. All optimized derivatives complied with Lipinskiʼs Rule of Five (0 violations), showed enhanced predicted antibacterial and membrane-disrupting activity scores, and maintained favorable toxicity profiles. Molecular docking revealed that modified compounds achieved substantially stronger predicted binding affinities than their native forms, with modified Baeckenone B showing the most promising profile (ΔG = ‒7.4 kcal/mol, Ki = 0.58 µM) exceeding the non-covalent affinity of fosfomycin (ΔG = ‒4.5 kcal/mol). Molecular dynamics simulations corroborated these findings, demonstrating superior complex stability (RMSD = 0.612 Å for modified Baeckenone B), reduced conformational fluctuations, and highly favorable MM/PBSA binding free energies (‒150 to ‒280 kcal/mol). Crucially, these computational predictions require experimental validation. The identified candidates, particularly modified Baeckenone B, should be synthesized and subjected to in vitro MurA enzymatic inhibition assays and antibacterial susceptibility testing against relevant pathogens. Only through such experimental confirmation can the true therapeutic potential of these baeckenone derivatives be established.
Acknowledgements
The authors would like to express their sincere gratitude to the OECD QSAR Toolbox server for providing valuable computational resources and support in conducting the QSAR analysis. The availability of this platform has significantly contributed to the completion of this research. This research was supported by the computational facilities of the Institute of Medical Education, Research, Service, and Innovation (IMERSI) Laboratory at the Faculty of Medicine at Universitas Pembangunan Nasional Veteran Jawa Timur. The authors extend their sincere gratitude to IMERSI for providing access to their high-performance computing (HPC) cluster, without which the molecular docking and dynamics simulations central to this study would not have been feasible.
Declaration of Generative AI in Scientific Writing
The authors affirm that the usage of generative artificial intelligence tools, including DeepL and ChatGPT (OpenAI), is intended solely for language refinement and grammatical correction throughout the manuscript drafting process. These tools were not used for generating content, data analysis, or interpretation. The authors are fully responsible for the accuracy and integrity of the content as well as the conclusions presented in this study.
CRediT Author Statement
Ilham Kurniawan: Conceptualization; Methodology; Software; Formal Analysis; Resources; Writing-Original Draft; Visualization. Desmila Idola: Conceptualization; Performed the experiment, Formal Analysis; Data curation; Writing-Original draft preparation; Visualization; Project Administration. Ippei Niwata: Performed the experiment, Formal Analysis; Data curation. Aulia Umi Rohmatika: Writing-Original Draft; Resources; Visualization; Investigation; Formal Analysis; Project Administration. I Made Artika: Supervision, Writing-Original draft preparation. Hisashi Muramatsu: Supervision, Writing-Original draft preparation. Chul-Sa Kim: Supervision, Writing-Original draft preparation. Takehiro Kashiwagi: Supervision, Writing-Original draft preparation. Wien Kusharyoto: Supervision. Fachrur Rizal Mahendra: Software; Resources; Data Curation.
References
[1] AW Septama, N Simbak and EP Rahmi. Prospect of plant-based flavonoids to overcome antibacterial resistance: A mini-review. Walailak Journal of Science and Technology 2020; 17(5), 503-513.
[2] M Song, Y Liu, T Li, X Liu, Z Hao, S Ding, P Panichayupakaranant, K Zhu and J Shen. Plant natural flavonoids against multidrug resistant pathogens. Advanced Science 2021; 8(15), 2100749.
[3] K Dwivedi, AK Mandal, O Afzal, ASA Altamimi, A Sahoo, MA Alossaimi, WH Almalki, A Alzahrani, MA Barkat, TM Almeleebia, SNMN Ullah and M Rahman. Emergence of nano-based formulations for effective delivery of flavonoids against topical infectious disorders. Gels 2023; 9(8), 671.
[4] F Farhadi, B Khameneh, M Iranshahi and M Iranshahy. Antibacterial activity of flavonoids and their structure–activity relationship: An update review. Phytotherapy Research 2018; 33(1), 13-40.
[5] M Miklasińska‐Majdanik, M Kępa, RD Wojtyczka, D Idzik and TJ Wąsik. Phenolic compounds diminish antibiotic resistance of Staphylococcus aureus clinical strains. International Journal of Environmental Research and Public Health 2018; 15(10), 2321.
[6] FA Resende, LG Nogueira, TM Bauab, W Vilegas and EA Varanda. Antibacterial potential of flavonoids with different hydroxylation patterns. Eclética Química 2015; 40, 173-179.
[7] T WY and B GD. Unusual metabolites of Baeckea frutescens. Tetrahedron 1996; 52(29), 9735-9742.
[8] K Nisa, T Ito, Subehan, T Matsui, T Kodama and H Morita. New acylphloroglucinol derivatives from the leaves of Baeckea frutescens. Phytochemistry Letters 2016; 15, 42-45.
[9] DTL Huong, DX Duc and NT Son. Baeckea frutescens L: A review on phytochemistry, biosynthesis, synthesis, and pharmacology. Natural Product Communications 2023; 18(7), 1-22.
[10] Y Zhou, B Utama, S Pratap, A Supandy, X Song, TT Tran, HH Mehta, CA Arias and Y Shamoo. Enolpyruvate transferase MurAAA149E, identified during adaptation of enterococcus faecium to daptomycin, increases stability of MurAA–MurG interaction. Journal of Biological Chemistry 2023; 299(3), 102912.
[11] A Sorlózano, I Lopez-Machado, M Albertuz-Crespo, LJ Martínez-González and JG Fernández. Characterization of fosfomycin and nitrofurantoin resistance mechanisms in Escherichia Coli isolated in clinical urine samples. Antibiotics 2020; 9(9), 534.
[12] AH Lima, AM dos Santos, CN Alves and J Lameira. Computed insight into a peptide inhibitor preventing the induced fit mechanism of MurA enzyme from Pseudomonas aeruginosa. Chemical Biology & Drug Design 2016; 89(4), 599-607.
[13] OB Kapitan, L Ambarsari and S Falah. Inhibition docking simulation of zerumbone, gingerglycolipid B, and curzerenone compound of zingiber zerumbet from timor Island against MurA enzyme. Journal of Applied Chemical Science Internationalpp 2016; 3, 279-288.
[14] S Kamboj and R Singh. Chromanone-A prerogative therapeutic scaffold: An overview. Arabian Journal for Science and Engineering 2022; 47(1), 75-111.
[15] P Petakh and O Kamyshnyi. AMR mechanisms in L. interrogans serovars: A comprehensive study. Frontiers in Cellular and Infection Microbiology 2024; 14, 1384427.
[16] FM Sezgin, M Avcu, E Sevim and UT Babaoğlu. In vitro activity of fosfomycin on biofilm in community-acquired staphylococcus aureus isolates. Clinical and Experimental Health Sciences 2019; 9(3), 202-209.
[17] Y Tanrıverdi-Çaycı, DB Güne, M Ertokatlı, K Hacıeminoğlu-Ülker and A Birinci. Prevalence of fosfomycin resistance among enterobacterales isolates in a tertiary care Hospital from Turkey. Infectious Diseases & Clinical Microbiology 2022; 4(4), 252-257.
[18] X Yu, X Zhu, Y Zhou, Q Li, Z Hu, T Li, J Tao, M Dou, M Zhang and Y Shao and R Sun. Discovery of N-Aryl-Pyridine-4-Ones as novel potential agrochemical fungicides and bactericides. Journal of Agricultural and Food Chemistry 2019; 67(50), 13904-13913.
[19] R Frlan, M Hrast and S Gobec. Inhibition of MurA enzyme from escherichia coli by flavonoids and their synthetic analogues. ACS Omega 2023; 8(36), 33006-33016.
[20] I Kurniawan, L Ambarsari, PA Kurniatin and ST Wahyudi. Novel compounds design of acertannin, hamamelitannin, and Petunidin-3-Glucoside typical compounds of african leaves (Vernonia amygdalina Del) as antibacterial based on QSAR and molecular docking. Jurnal Jamu Indonesia 2023; 8(2), 326.
[21] I Kurniawan and NS Winarno. Unlocking antibacterial potential: Thiophene-2-carbaldehyde modification of acertannin from african leaves as MurA enzyme inhibitors. Jurnal Ners 2025; 9(4), 7602-7612.
[22] SA Khan, SU Khan, Fozia, N Ullah, M Shah, R Ullah, I Ahmad and A Alotaibi. Isolation, structure elucidation and in silico prediction of potential drug-like flavonoids from Onosma chitralicum targeted towards functionally important proteins of drug-resistant bad bugs. Molecules 2021; 26(7), 2048.
[23] BD VanScoy, J McCauley, EJ Ellis-Grosse, OO Okusanya, SM Bhavnani, A Forrest and PG Ambrose. Exploration of the pharmacokinetic-pharmacodynamic relationships for fosfomycin efficacy using an in vitro infection model. Antimicrobial Agents and Chemotherapy 2015; 59(12), 7170-7177.
[24] AAN Shadrina, Y Herdiyati, I Wiani, MH Satari and D Kurnia. Prediction mechanism of nevadensin as antibacterial agent against S. Sanguinis: In vitro and in silico studies. Combinatorial Chemistry & High Throughput Screening 2022; 25(9), 1488-1497.
[25] Q Liu, A Luo, H Jin, X Si and M Li. Machine learning‐based discovery of a novel noncovalent MurA inhibitor as an antibacterial agent. Chemical Biology & Drug Design 2025; 105(3), 70084.
[26] JP James, PD Ail, L Crasta, RS Kamath, MH Shura and TJ Sindhu. In silico ADMET and molecular interaction profiles of phytochemicals from medicinal plants in Dakshina Kannada. Journal of Health and Allied Sciences NU 2024; 14, 190-201.
[27] J Wang, F Yuan, M Kendre, Z He, S Dong, A Patil and K Padvi. Rational design of allosteric inhibitors targeting C797S mutant EGFR in NSCLC: An integrative in silico and in-vitro study. Frontiers in Oncology 2025; 15, 1590779.
[28] B Li, Z Wang, Z Liu, Y Tao, C Sha, M He and X Li. DrugMetric: Quantitative drug-likeness scoring based on chemical space distance. Briefings in Bioinformatics 2024; 25(4), 321.
[29] X Chen, H Li, L Tian, Q Li, J Luo and Y Zhang. Analysis of the physicochemical properties of acaricides based on Lipinski’s rule of five. Journal of Computational Biology 2020; 27(9), 1397-1406.
[30] C Boss, J Hazemann, T Kimmerlin, M von Korff, U Lüthi, O Peter, T Sander and R Siegrist. The screening compound collection: A key asset for drug discovery. CHIMIA 2017; 71, 667.
[31] K Long, SJ Kostman, CA Fernandez, JC Burnett and DM Huryn. Do zebrafish obey lipinski rules? ACS Medicinal Chemistry Letters 2019; 10(6), 1002-1006.
[32] A Furukawa, CE Townsend, J Schwochert, CR Pye, MA Bednarek and RS Lokey. Passive membrane permeability in cyclic peptomer scaffolds is robust to extensive variation in side chain functionality and backbone geometry. Journal of Medicinal Chemistry 2016; 59(20), 9503-9512.
[33] A Singh and MA Azam. MurF ligase inhibitors: An overview of antibacterial activity. Letters in Drug Design & Discovery 2023; 20(11), 1675-1687.
[34] I Kurniawan and AU Rohmatika. Molecular docking, QSAR, and bioactivity prediction of Uncaria gambir flavonoids as antibacterial agents targeting MurA enzyme. Biointerface Research in Applied Chemistry 2026; 16(1), 32.
[35] A Hentsch, M Guberman, S Radetzki, S Kaushik, M Huizenga, Y He, J Contzen, B Kuhn, J Benz, M Schippers, J Paul, L Leibrock, L Collin, M Wittwer, A Topp, F O’Hara, D Heer, R Hochstrasser, J Blaising, JP von Kries and M Nazaré. Highly specific miniaturized fluorescent monoacylglycerol lipase probes enable translational research. Journal of the American Chemical Society 2025; 147(12), 10188-10202.
[36] A Kousaxidis, L Kováčiková, I Nicolaou, M Štefek and A Geronikaki. Non-acidic bifunctional benzothiazole-based thiazolidinones with antimicrobial and aldose reductase inhibitory activity as a promising therapeutic strategy for sepsis. Medicinal Chemistry Research 2021; 30, 1837-1848.
[37] Z Ranković. CNS drug design: Balancing physicochemical properties for optimal brain exposure. Journal of Medicinal Chemistry 2015; 58(6), 2584-2608.
[38] Y Goto and H Suga. The RaPID platform for the discovery of pseudo-natural macrocyclic peptides. Accounts of Chemical Research 2021; 54(18), 3604-3617.
[39] R Mutiah, C Ayatillah, YYA Indrawijaya and A Suryadinata. Prediction of anti-sars CoV-2 activity from green tea catechin (Camellia Sinensis L. Kuntze) compound against to receptors non-structural protein 3 (6W6Y) and non-structural protein 5 (6M2N). Majalah Obat Tradisional 2022; 27(1), 24-31.
[40] S Atoyebi, F Bunglawala, N Cottura, S Grañana-Castillo, MC Montanha, A Olagunju, M Siccardi and C Waitt. Physiologically‐based pharmacokinetic modelling of long‐acting injectable cabotegravir and rilpivirine in pregnancy. British Journal of Clinical Pharmacologyvol 2024; 91(4), 989-1002.
[41] AP Sousa, MS Oliveira, DA Fernandes, MDL Ferreira, LV Cordeiro, MFV Souza, LMD Fernandes, HDS Souza, AAO Filho, HLF Pessoa and RCS Sá. In silico, in vitro, and ex vivo studies of the toxicological and pharmacological properties of the flavonoid 5,7-Dihydroxy-3,8,4’-Trimethoxy. Brazilian Journal of Medical and Biological Research 2021; 54(10), 11203.
[42] JTS Coimbra, R Feghali, RP Ribeiro, MJ Ramos and PA Fernandes. The importance of intramolecular hydrogen bonds on the translocation of the small drug piracetam through a lipid bilayer. RSC Advances 2020; 11(2), 899-908.
[43] SH Emam, RA Hassan, EO Osman, MIA Hamed, AM Abdou, MM Kandil, EM Elbaz and DS Mikhail. Coumarin derivatives with potential anticancer and antibacterial activity: Design, synthesis, VEGFR‐2 and DNA gyrase inhibition, and in silico studies. Drug Development Research 2023; 84(3), 475-499.
[44] S Gavale, D Zade, KB Patil, R Yadav, PR Murumkar and R Kadu. Synthesis, characterization, and computational insights of flavone derivatives as promising antimicrobial agents. Chemistry & Biodiversity 2025; 22(12), 02136.
[45] MRU Ahsan, S Paul, MS Alam and AFMM Rahman. Synthesis, biological properties, in silico ADME, molecular docking studies, and FMO analysis of chalcone derivatives as promising antioxidant and antimicrobial agents. ACS Omega 2025; 10(5), 4367-4387.
[46] SU Chavan, SA Waghmare, SS Bodke and AR Bendale. Exploring 1,3,4-Oxadiazole derivatives as potent α-amylase inhibitors: Design, synthesis, and biological evaluation. Eurasian Journal of Chemistry 2023; 28(4), 112.
[47] A Chaurasyia, PA Chawla, V Monga and G Singh. Rhodanine derivatives: An insight into the synthetic and medicinal perspectives as antimicrobial and antiviral agents. Chemical Biology & Drug Design 2022; 101(3), 500-549.
[48] Y Zhou, B Utama, S Pratap, A Supandy, X Song, TT Tran, HH Mehta, CA Arias and Y Shamoo. Enolpyruvate transferase MurAAA149E, identified during adaptation of enterococcus faecium to daptomycin, increases stability of MurAA–MurG interaction. Journal of Biological Chemistryvol 2023; 299(3), 102912.
[49] SH Pattanashetty, KM Hosamani, AK Shettar and RM Shafeeulla. Design, synthesis and computational studies of novel carbazole n‐phenylacetamide hybrids as potent antibacterial, anti‐inflammatory, and antioxidant agents. Journal of Heterocyclic Chemistry 2018; 55(7), 1765-1774.
[50] I Kurniawan and AU Rohmatika. Molecular docking, QSAR, and bioactivity prediction of Uncaria gambir flavonoids as antibacterial agents targeting MurA enzyme. Biointerface Research in Applied Chemistry 2026; 16(1), 32.
[51] Jia, M Zheng, C Zhang, B Li, C Lu, Y Bai, Q Tong, X Hang, Y Ge, L Zeng, M Zhao, F Song, H Zhang, L Zhang, K Hong and H Bi. Killing of Staphylococcus aureus persisters by a multitarget natural product chrysomycin A. Science Advances 2023; 9(31), 5995.
[52] I Kurniawan and H Zahra. Review: Gallotannins; biosynthesis, structure activity relationship, anti-inflammatory and antibacterial activity. Current Biochemistry 2021; 8(1), 1-16.
[53] I Purwaningsih, IP Maksum, D Sumiarsa and S Sriwidodo. Pharmacokinetics and toxicity overview of active compounds berberine, palmatine, and jatrorrhizine from fibraurea tinctoria lour: Drug-likeness, ADMET prediction, and in vivo extract toxicity assessment. Journal of Toxicology 2025; 7251602.
[54] J Bradic, A Petrovic, M Nikolic, N Nedeljkovic, M Andjic, N Kladar, S Bolevich, V Jakovljevic and A Kocovic. Newly developed semi-solid formulations containing Mellilotus officinalis extract: Characterization, assessment of stability, safety, and anti-inflammatory activity. Pharmaceutics 2024; 16(8), 1003.
[55] NT El‐Shamy, A Alkaoud, RK Hussein, MA Ibrahim, AG Alhamzani and MM Abou-Krisha. DFT, ADMET and molecular docking investigations for the antimicrobial activity of 6,6′-Diamino-1,1′,3,3′-tetramethyl-5,5′-(4-chlorobenzylidene)bis[pyrimidine-2,4(1H,3H)-dione]. Molecules 2022; 27(3), 620.
[56] PS Ramalingam, P Balakrishnan, S Rajendran, J Arunachalam, R Rajasekaran and S Arumugam. Identification of dietary bioflavonoids as potential inhibitors against KRAS G12D mutant—novel insights from computer-aided drug discovery. Current Issues in Molecular Biology 2023; 45(3), 2136-2156.
[57] O Ejiohuo, D Bajia, J Pawlak and A Szczepankiewicz. Asoprisnil as a novel ligand interacting with stress-associated glucocorticoid receptor. Biomedicines 2024; 12(12), 2745.
[58] Y Sun, M Zhang, Y Zhang, Y Zheng, J Li, Q Cai, A Wang and Y Qu. Synergistic and antagonistic mechanisms of Arctium Lappa L. polyphenols on human neutrophil elastase inhibition: Insights from molecular docking and enzymatic kinetics. Molecules 2025; 30(13), 2764.
[59] A Dubey, S Dotolo, PW Ramteke, A Facchiano and A Marabotti. Searching for chymase inhibitors among chamomile compounds using a computational-based approach. Biomolecules 2019; 9(1), 5.
[60] M Islam, A Hossain, I Yamari, O Abchir, S Chtita, F Ali, SMA Kawsar. Synthesis, antimicrobial, molecular docking against bacterial and fungal proteins and in silico studies of glucopyranoside derivatives as potent antimicrobial agents. Chemistry & Biodiversity 2024; 21(9), 202400932.
[61] S Salentin, SB Schreiber, VJ Haupt, MF Adasme and M Schroeder. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Research 2015; 43(1), 443-447.
[62] MY Jin, H Yu, Q Deng, Z Wang, JY Wang, HL Li and H Liang. Virtual screening and molecular dynamics simulation study of ATP-competitive inhibitors targeting mTOR protein. PLoS One 2025; 20(5), 0319608.
[63] A Musoev, S Numonov, Z You and H Gao. Discovery of novel DPP-IV inhibitors as potential candidates for the treatment of Type 2 Diabetes mellitus predicted by 3D QSAR pharmacophore models, molecular docking and de novo evolution. Molecules 2019; 24(16), 2870.
[64] L Zheng, J Fan and Y Mu. OnionNet: A multiple-layer intermolecular-contact-based convolutional neural network for protein–ligand binding affinity prediction. ACS Omega 2019; 4(14), 15956-15965.
[65] D Dubey, A Dubey, A Tufail and AK Rai. Assessment of optimal analytical techniques and computational approaches for investigating toxic elements and compounds in pyrotechnic materials (Green Crackers). Applied Organometallic Chemistry 2025; 39(9), 70361.
[66] D Solano-Orrala, DA Silva-Cullishpuma, E Díaz-Cruces, VM Gómez-López, J Toro-Mendoza, GG D’Ayala, J Troconis, C Narváez-Muñoz, F Alexis, MT Mercader-Ros, C Lucas-Abellán and C Zamora-Ledezma. Exploring the potential of nonpsychoactive cannabinoids in the development of materials for biomedical and sports applications. ACS Applied Bio Materials 2024; 7(12), 8177-8202.
[67] MF Chabán, M Hrast, R Frlan, DG Graikioti, CM Athanassopoulos and MC Carpinella. Inhibition of MurA enzyme from Escherichia coli and Staphylococcus aureus by diterpenes from Lepechinia meyenii and their synthetic analogs. Antibiotics 2021; 10(12), 1535.
[68] MVD de Oliveira, KS da Costa, JRA Silva, J Lameira and AH Lima. Role of UDP‐N‐acetylmuramic acid in the regulation of mura activity revealed by molecular dynamics simulations. Protein Science 2024; 33(4), 4969.
[69] ES İstifli. Chemical composition, antioxidant and enzyme inhibitory activities of Onosma bourgaei and Onosma trachytricha and in silico molecular docking analysis of dominant compounds. Molecules 2021; 26(10), 2981.
[70] FR Mahendra, I Prakoso, A Marzelino, MR Fadhillah, Syukriyansyah, MMA Hasibuan, JF Suparningtyas, M Kristiadi, AG Setiawan, FI Rahman, AN Sari, NR Raysendria, I Kurniawan, W Arozal and Kusmardi. Optimisation of peptides targeting reverse transcriptase HIV-1 using QSAR, machine learning, and computational approaches. Frontiers in Pharmacology 2025; 16, 1707377.
[71] RAP Irsal, GM Gholam, DA Firdaus, N Liwanda and F Chairunisa. Molecular docking and dynamics of xylocarpus granatum as a potential parkinson’s drug targeting multiple enzymes. Borneo Journal of Pharmacy 2024; 7(2), 161-171.
[72] F Zhang, J Graham, T Zhai, Y Liu and Z Huang. Discovery of MurA inhibitors as novel antimicrobials through an integrated computational and experimental approach. Antibiotics (Basel, Switzerland) 2022; 11(4), 528.
[73] MT Khazaal, AHI Faraag and HH El-Hendawy. In vitro and in silico studies of enterobactin-inspired ciprofloxacin and fosfomycin first generation conjugates on the antibiotic resistant E. Coli OQ866153. BMC Microbiology 2024; 24, 95.
[74] S Akash, G Abdelkrim, I Bayil, ME Hosen, N Mukerjee, AF Shater, FM Saleh, GM Albadrani, MQ Al-Ghadi, MM Abdel-Daim and TT Tok. Antimalarial drug discovery against malaria parasites through haplopine modification: An advanced computational approach. Journal of Cellular and Molecular Medicine 2023; 27(20), 3168-3188.
[75] MLBEN Amor, E Lanez, Y Bekkar, A Adaika, T Lanez, K Nesba and L Bechki. Exploring the interactions of ferrocenylmethylnucleobase derivatives with BSA and HHb: Insights from electrochemical, spectroscopic, ADMET, in silico docking, and MD simulations. ChemistrySelect 2025; 10(6), 202404678.
[76] JU Ting, MCS Tan, VAS Ng, SJY Macalino, VC Linis and GG Oyong. Molecular simulations of unexplored philippine plant constituents on the inhibition of the proinflammatory marker NF-κB P50 subunit. Crystals 2024; 14(5), 438.
[77] AK Umar, JH Zothantluanga, K Aswin, S Maulana, MS Zubair, H Lalhlenmawia, M Rudrapal and D Chetia. Antiviral phytocompounds “ellagic acid” and “(+)-sesamin” of bridelia retusa identified as potential inhibitors of SARS-CoV-2 3CL pro using extensive molecular docking, molecular dynamics simulation studies, binding free energy calculations, and bioactivi. Structural Chemistry 2022; 33, 1445-1465.
[78] MH Abbas, AR Alanzi, KI Sahibzada, M Nawaz, G Fatima and D Wei. Identification of novel inhibitors targeting Mycobacterium abscessus InhA through virtual screening, docking, and molecular dynamic simulations. Scientific Reports 2025; 15, 12795.
[79] SJ Hamid, T Salih and TA Aziz. Rational design, computational analysis and antibacterial activities of synthesized peptide-based molecules targeting quorum sensing-dependent biofilm formation in Pseudomonas aeruginosa. Pharmaceuticals 2025; 18(10), 1572.
[80] M Yasir, J Park, ET Han, JH Han, WS Park, J Choe and W Chun. Investigating natural product inhibitors of IKKα: Insights from integrative in silico and experimental validation. Molecules 2025; 30(9), 2025.
[81] M Menteş and C Yandım. Identification of PPA1 inhibitor candidates for potential repurposing in cancer medicine. Journal of Cellular Biochemistry 2023; 124(10), 1646-1663.
[82] RM Laki and E Pourbasheer. Design of new anti‐influenza structures based on 3D‐QSAR, molecular docking and molecular dynamics studies. Chemistry & Biodiversity 2025; 22(9), 202500587.
[83] F Zare, E Ataollahi, P Mardaneh, A Sakhteman, V Keshavarz, A Solhjoo and L Emami. A combination of virtual screening, molecular dynamics simulation, MM/PBSA, ADMET, and DFT calculations to identify a potential DPP4 inhibitor. Scientific Reports 2024; 14, 7749.
[84] NH Aljarba, MS Hasnain, MM Bin-Meferij and S Alkahtani. Design & discovery of small molecule COVID-19 inhibitor via dual approach based virtual screening and molecular simulation studies. Journal of King Saud University - Science 2022; 34, 101867.
[85] BJ Harris, X Cheng and PD Frymier. Structure and function of photosystem I–[FeFe] Hydrogenase protein fusions: An all-atom molecular dynamics study. The Journal of Physical Chemistry B 2016; 120(4), 599-609.
[86] KM Gokhale, N Joshi and RR Alavala. Acetohydroxyacid Synthase (AHAS) inhibitors as antitubercular agents: Insights from molecular docking and dynamics simulations. Chemistry & Biodiversity 2025; 22(6), 202402631.
[87] AA Hendi, P Virk, MA Awad, M Elobeid, KMO Ortashi, MM Alanazi, FH Alkallas, MM Almoneef and MA Abdou. In silico studies on zinc oxide based nanostructured oil carriers with seed extracts of Nigella sativa and Pimpinella anisum as potential inhibitors of 3CL protease of SARS-CoV-2. Molecules 2022; 27(13), 4301.
[88] S Ullah, F Ullah, W Rahman, A Ullah, S Haider and C Yueguang. Elucidating the inhibitory mechanism of Zika Virus NS2B-NS3 protease with dipeptide inhibitors: Insights from molecular docking and molecular dynamics simulations. PLoS One 2024; 19(8), 0307902.