Trends
Sci.
2026; 23(5):
12146
In silico and In vitro Activity of Leonurus turkestanicus on the Aortic Smooth Muscle Ion Channels
Gulhayoxon Mirzaalimova1,*, Anvar Zaynabiddinov1, Sirojiddin Omonturdiev2,
Dolimjon Inomjonov3, Izzatullo Abdullaev2, Ulugbek Gayibov2 and Madina Kholmirzayeva1
1Department of Human Physiology and Life Safety, Andijan State University, Andijan 170100, Uzbekistan
2A. S. Sadykov Institute of Bioorganic Chemistry of the Science Academy of Uzbekistan, Tashkent 100143, Uzbekistan
3Department of Anatomy and Physiology, Namangan State University, Namangan 160107, Uzbekistan
(*Corresponding author’s e-mail: [email protected])
Received: 2 October 2025, Revised: 19 October 2025, Accepted: 5 November 2025, Published: 5 January 2026
Abstract
Leonurus turkestanicus is traditionally used for cardiovascular disorders, yet its phytochemical profile and vasorelaxant mechanisms remain insufficiently characterized. In this study, the aqueous extract of L. turkestanicus was analyzed by high-performance liquid chromatography (HPLC), which revealed a rich composition of flavonoids and phenolic acids, including rutin, isoquercitrin, hyperoside, quercetin, apigenin, gallic acid, p-coumaric acid, and ferulic acid. Functional assays in isolated rat aortic rings demonstrated that the extract produced concentration-dependent vasorelaxation against KCl- and phenylephrine-induced contractions. These effects were mediated through inhibition of voltage-dependent L-type calcium channels and receptor-operated calcium channels, as well as through endothelium-dependent nitric oxide signaling. The vasorelaxant action was further supported by additive interactions with verapamil and phentolamine. Molecular docking analyses confirmed that major flavonoids strongly interact with multiple calcium-handling proteins, including L-type and R-type calcium channels, NCX1, SERCA and RyR2, with rutin and hyperoside exhibiting the highest inhibitory affinities. Collectively, these findings indicate that L. turkestanicus exerts vasoprotective effects through a multi-targeted modulation of calcium signaling and endothelial function. The study provides mechanistic evidence supporting the traditional use of L. turkestanicus and identifies its bioactive flavonoids as promising leads for antihypertensive drug development.
Keywords: Leonurus turkestanicus, Phytochemical profiling, Flavonoids, Calcium channels, Vasorelaxation, Nitric oxide, Molecular docking, Antihypertensive activity
Introduction
Hypertension remains one of the leading global health challenges, contributing substantially to cardiovascular morbidity and mortality. Although a wide range of antihypertensive drugs are available, their long-term administration is often accompanied by adverse effects, tolerance, and limited efficacy in specific patient populations. These limitations have intensified the search for natural, plant-derived compounds as complementary or alternative therapeutic strategies. Phytochemicals, particularly flavonoids and phenolic acids, have been widely recognized for their antioxidant, anti-inflammatory, and vasodilatory properties that collectively contribute to blood pressure reduction [1,2].
Voltage-gated calcium (Ca²⁺) channels and other ion transport systems in vascular smooth muscle cells play a central role in regulating vascular tone and peripheral resistance. Modulation of these calcium-handling pathways represents one of the main therapeutic strategies for antihypertensive agents [3,4]. Although numerous medicinal plants have been reported to exert vascular benefits, the specific molecular mechanisms—especially their direct interactions with calcium transport proteins—remain poorly characterized [5].
Within this context, the genus Leonurus (family Lamiaceae) includes several species traditionally used for cardiovascular disorders, such as Leonurus cardiaca and Leonurus japonicus, both of which have demonstrated cardiotonic, antioxidant, and vasodilatory properties. However, the Central Asian endemic species Leonurus turkestanicus has received comparatively little scientific attention despite its extensive ethnopharmacological use in the treatment of palpitations, hypertension, and nervous system disorders. Preliminary phytochemical studies suggest that L. turkestanicus contains a distinct flavonoid profile, characterized by higher concentrations of rutin, hyperoside, and isoquercitrin, which differentiates it from other Leonurus species and may underlie its unique pharmacological properties.
Therefore, the present study aimed to investigate the vascular and molecular mechanisms of L. turkestanicus aqueous extract, with a particular focus on its modulation of calcium transport systems in vascular smooth muscle. By combining in vitro vascular reactivity assays with in silico molecular docking analyses, this research sought to elucidate how the plant’s bioactive constituents influence voltage- and receptor-operated calcium channels, Ca²⁺-ATPases, and other calcium-handling proteins. Through this integrative approach, the study provides new insights into the mechanistic basis of the antihypertensive and vasoprotective effects of L. turkestanicus, filling an important gap in the understanding of calcium-mediated actions of Leonurus species.
Materials and methods
Plant material and extraction
The Leonurus turkestanicus V.I. Krecz [6,7]. extract used in this study was kindly provided by Bioton LTD (Tashkent, Uzbekistan). The plant material was collected from natural populations in Uzbekistan and authenticated by specialists of the company. The dried aerial parts were subjected to ethanol extraction under standardized conditions, followed by filtration and solvent evaporation to obtain a concentrated dry extract. The extract was stored in airtight containers at 4 °C until further use. For experimental procedures, stock solutions were freshly prepared in distilled water or physiological saline, depending on the assay requirements (Figure 1).

Figure 1 The Leonurus turkestanicus V.I. Krecz.
Chemicals
Acetonitrile, Phenylephrine, verapamil (≥98% purity) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
High-Performance Liquid Chromatography (HPLC) analysis
The phytochemical composition of the extract was analyzed using High-Performance Liquid Chromatography (HPLC). For sample preparation, 1 mg of the powdered extract was weighed and dissolved in 1 mL of 25% acetonitrile. The mixture was sonicated for 3 min, followed by centrifugation [8,9]. The supernatant was filtered through a 0.45 μm membrane filter and used for HPLC injection. Chromatographic analysis was performed using an Agilent 1,260 HPLC system equipped with an autosampler. Separation was achieved on a Poroshell 120 EC-C18 column (4 μm, 4.6×250 mm). The column temperature was maintained at 30 °C, and the flow rate was set at 0.9 mL/min. Detection wavelengths were 254, 269 and 226 nm. The mobile phase consisted of solvent C (acetonitrile) and solvent D (0.1% trifluoroacetic acid buffer, pH = 3.0). A gradient elution program was applied as follows: 0 - 2 min, 15% acetonitrile (v/v); 2 - 42 min, 42% acetonitrile (v/v); 42 - 46 min, 15% acetonitrile (v/v). Prior to sample analysis, standard reference solutions were injected under the same chromatographic conditions.
Animal ethics
All preoperative and experimental procedures were carefully reviewed and approved by the Institutional Committee for Animal Use and Care. The animals were housed in a vivarium under standardized conditions, including a relative humidity of 55% - 65%, a controlled ambient temperature of 22 ± 2°C, and unrestricted access to water and standard laboratory chow. All aspects of animal care and handling were conducted in full compliance with the European Directive 2010/63/EU, which governs the protection of animals used for scientific research. Ethical clearance for this study was granted by the Animal Ethics Committee of the Institute of Bioorganic Chemistry, Academy of Sciences of the Republic of Uzbekistan (Protocol No. 133/1a/h, dated 4 August 2016).
Tissue preparation
All surgical procedures were carried out under sodium pentobarbital anesthesia to ensure the animals remained free of pain. Thoracic aortas were obtained from healthy adult male Wistar rats weighing 200 - 250 g. Animals were euthanized by cervical dislocation, after which a thoracotomy was performed to excise the aorta. Perivascular adipose and connective tissues were carefully removed, and the vessels were cut into rings approximately 3 - 4 mm in length. These aortic segments were placed in a 5 mL organ bath containing Krebs–Henseleit buffer (in mM: NaCl 120.4, KCl 5, NaHCO₃ 15.5, NaH₂PO₄ 1.2, MgCl₂ 1.2, CaCl₂ 2.5, glucose 11.5 and HEPES; pH 7.4). For selected experiments, a calcium-free Krebs solution containing 1 mM EGTA was used to evaluate calcium-dependent contractile responses. The buffer was continuously aerated with carbogen gas (95% O₂, 5% CO₂) and maintained at 37 °C using a DAIHAN ultrathermostatic water bath [10,11].
Aortic-ring contraction studies
Isolated aortic rings were mounted in a Radnoti isometric transducer system (USA) using platinum wire hooks and allowed to equilibrate for 60 min under physiological conditions (Figure 2). Each ring was subjected to a resting preload of 1 g (≈10 mN). Contractile activity was recorded with an isometric force transducer connected to a signal amplifier, and data were digitized using a Go-Link analog-to-digital converter interfaced with a computer. Data collection and analysis were performed with Origin Pro software, version 9 SR1 (EULA, Northampton, MA, USA). Contractile responses were expressed as isometric tension (mN) relative to the maximal contraction [12,13]. The functional integrity of vascular smooth muscle preparations was confirmed following the protocol. The 5 mL organ bath was incorporated into a recirculating Krebs–Henseleit buffer system, maintained at 37 °C with a thermostatic water bath, and continuously aerated with carbogen (95% O₂, 5% CO₂). Isometric force was continuously monitored using a Grass Instruments force transducer (USA), with amplification and data acquisition achieved via the GoLink system [14].
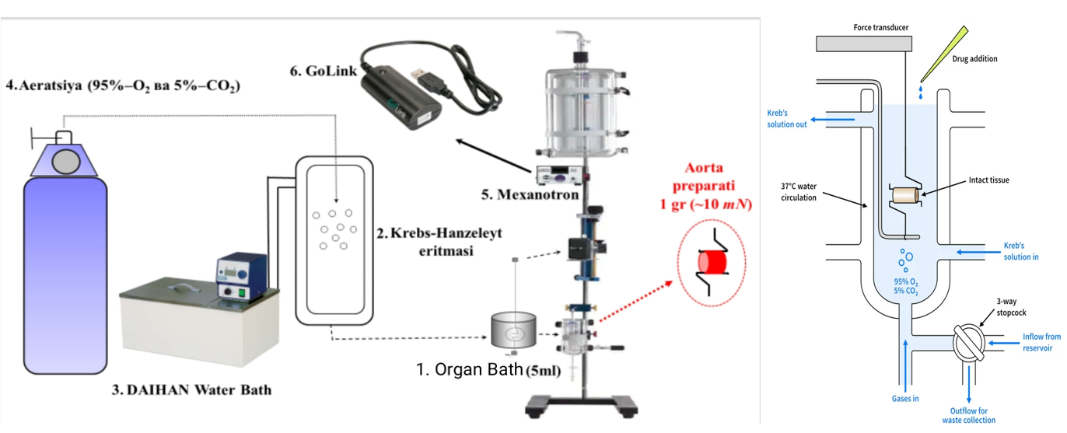
Figure 2 Diagram of the experimental setup used to monitor isometric contractions in isolated rat aortic vascular tissue. (1) The organ bath (5 mL) is connected to a dedicated reservoir for solution circulation. (2) Krebs-Henseleit solution is used to maintain physiological conditions. (3) A thermostat ensures stable temperature regulation. (4) The system is continuously aerated with a gas mixture containing 95% oxygen and 5% carbon dioxide. The aortic tissue is mounted within the experimental chamber for contraction assessment. (5) An isometric transducer (Grass Instrument, USA) captures mechanical responses, while (6) GoLink devices handle signal amplification and system integration.
Statistics
Data analysis and graph generation were performed using Origin Pro software, version 9 (USA). Vascular contractions were expressed as a percentage of the maximal response elicited by phenylephrine (10 mM) or potassium chloride (50 mM). Results are presented as mean ± standard error (SEM) from 4 - 6 independent experiments (n = 4 - 6). Paired Student’s t-tests were used to assess differences within the same group, whereas unpaired t-tests were applied for comparisons between groups. Statistical significance was defined as p < 0.05.
Molecular docking “Software and databases”
All computational tools applied in this study were freely accessible for academic and educational purposes. Structural data of macromolecules involved in calcium signaling and regulation were retrieved from the Protein Data Bank (PDB), an internationally recognized repository of three-dimensional biomolecular structures [15,16]. The chosen target proteins included the L-type calcium channel Cav1.2 (PDB ID: 6jp5), R-type calcium channel Cav2.3 (PDB ID: 7xlq), sodium–calcium exchanger NCX1 (PDB ID: 8sgi), ryanodine receptor type 2 RyR2 (PDB ID: 5c33), and sarcoplasmic/endoplasmic reticulum Ca²⁺-ATPase (SERCA, PDB ID: 6rb2). Reference compounds along with the flavonoid ligands of interest were obtained from the PubChem database, which provides extensive information on pharmacology, molecular targets, chemical structures, and biological pathways. Each PubChem DrugCard contains over 80 fields describing small molecules and their associated protein targets (Table 1). Molecular structures and docking results were visualized with PyMOL (version 1.2), a Python-based molecular graphics system [http://www.pymol.org]. Docking simulations were carried out using AutoDock 4.2, developed at The Scripps Research Institute (www.scripps.edu). Input preparation and parameter configuration were performed with AutoDock Tools (ADT), a user-friendly graphical interface designed to set up and run docking experiments. AutoDock provides a robust computational framework for predicting ligand–target binding conformations and interactions when three-dimensional structural data are available [17].
Calculation of inhibition constant (Ki) from binding energy.
In molecular docking studies, the interaction strength between a ligand and its target protein is commonly expressed as the binding free energy (ΔG), measured in kilocalories per mole (kcal/mol) [18]. This value can be further applied to estimate the inhibition constant (Ki), which reflects the ligand’s binding affinity, using a simple thermodynamic relationship.

where: Ki is the inhibition constant (in mol/L), ΔG is the binding free energy (in kcal/mol), R is the universal gas constant = 1.987 cal/(mol·K), T is the temperature in Kelvin (usually 298.15 K), The factor 1,000 converts kcal to cal.
Results
Phytochemical profiling of Leonurus turkestanicus extract by HPLC
The phytochemical composition of the aqueous extract of Leonurus turkestanicus was investigated using high-performance liquid chromatography (HPLC) under optimized gradient elution conditions. The applied gradient system, consisting of acetonitrile and 0.1% trifluoroacetic acid buffer, enabled effective separation of phenolic acids and flavonoids within 46 min of analysis [19].
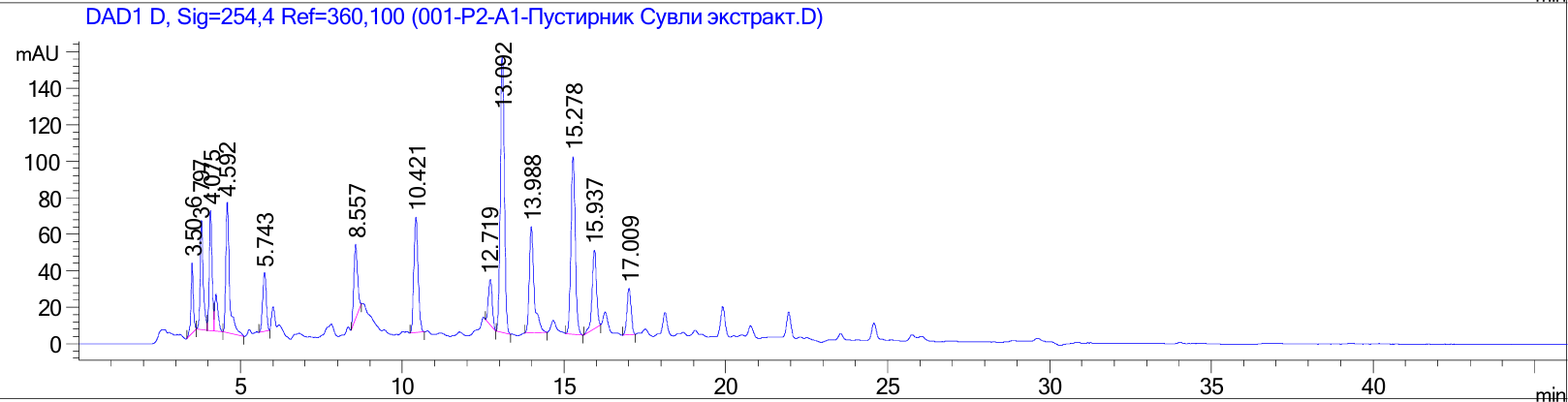
Figure 3 Graphical representation of the high-performance liquid chromatography result of the chemical composition of the extract.
The obtained chromatogram revealed multiple well-resolved peaks corresponding to different phenolic compounds (Figure 3). Identification of bioactive constituents was performed by comparing retention times and UV spectra with those of authentic reference standards. This comparative profiling confirmed the presence of diverse phytochemicals, mainly flavonoids and phenolic acids, within the extract.
The results demonstrated that the extract contained a rich array of compounds, including gallic acid, isorhamnetin, (+)-catechin, rutin, isoquercitrin (quercetin-3-O-glucoside), hyperoside (quercetin-3-D-galactoside), p-coumaric acid, quercetin, apigenin, and ferulic acid (Table 1). Among them, flavonoid glycosides such as rutin, isoquercitrin, and hyperoside were particularly abundant, suggesting their potential role as major contributors to the biological activities of the extract. Phenolic acids such as gallic acid, p-coumaric acid, and ferulic acid were also detected, providing evidence of the antioxidant potential of the extract.
Table 1 Compounds present in the extract.
Compounds present in the sample |
|||||
Retention time Rt, min |
Identified compounds |
Area% |
[MМ] |
Brutto formula |
Concentration mg/g |
3.563 |
Gallic acid |
3.093 |
176 |
C6H8O6 |
2.093 |
4.075 |
Isorhamnetin |
6.179 |
316 |
C16H12O7 |
5.89 |
5.74 |
+catechin |
3.75 |
290 |
C15H14O6 |
3.57 |
8.557 |
Quercetin-3-O-glucoside (Isoquercitrin) |
5.009 |
464 |
C21H20O12 |
4.77 |
10.421 |
Rutin |
8.885 |
610 |
C27H30O16 |
8.47 |
12.719 |
Hyperoside (Quercetin 3-D-galactoside) |
3.102 |
464 |
C21H20O12 |
2.95 |
13.988 |
P-coumaric acid |
20.074 |
C4H4O4 |
C4H4O4 |
19.14 |
15.278 |
Quercetin |
9.38 |
302 |
C15H10O7 |
8.94 |
18.346 |
Apigenin |
2.47 |
270 |
C15H10O5 |
2.35 |
19.78 |
Ferulic acid |
2.73 |
194 |
C10H10O4 |
2.6 |
Involvement of L-type and receptor-operated Ca²⁺ channels in the vasorelaxant action of L. turkestanicus
It is well recognized that 50 mM KCl induces contraction of aortic smooth muscle mainly through activation of voltage-dependent L-type calcium (Ca²⁺) channels [15]. An increase in extracellular potassium concentration causes membrane depolarization, which in turn alters the membrane potential and opens Ca²⁺ channels, facilitating calcium influx and initiating vasoconstriction. In the present study, the vasorelaxant effects of L. turkestanicus extract were evaluated on KCl-induced contractions in isolated rat aortic rings. The extract produced a concentration-dependent relaxation of pre-contracted vascular tissues. At 10 µg/mL, L. turkestanicus markedly suppressed 50 mM KCl-induced contractions, reducing contractile force from 3.0 ± 2.1% to 86.8 ± 2.5% relative to the control (Figure 4(A)). These observations suggest that the extract interferes with depolarization-induced calcium influx, most likely via modulation or blockade of L-type Ca²⁺ channels in vascular smooth muscle. By limiting extracellular calcium entry, which is essential for contraction, the extract lowers intracellular calcium levels and thereby facilitates vascular relaxation [20].
To further clarify the role of L-type calcium channels, verapamil—a standard L-type Ca²⁺ channel blocker—was used for comparison. Co-application of L. turkestanicus with a submaximal concentration of verapamil (0.1 μM), which only partially inhibited KCl-induced contractions, produced an additional 13.71 ± 3.5% relaxation, indicating an additive or possibly synergistic interaction (Figure 4(C)). The half-maximal inhibitory concentration (IC₅₀) for L. turkestanicus was estimated to be 40.97 µg/mL, underscoring its pharmacological activity.
Collectively, these findings demonstrate that L. turkestanicus mediates vasorelaxation primarily through inhibition of voltage-gated L-type Ca²⁺ channels, thereby reducing calcium influx and promoting smooth muscle relaxation. The overlap of its mechanism with that of verapamil points to the therapeutic potential of L. turkestanicus in vascular disorders characterized by calcium channel overactivity.
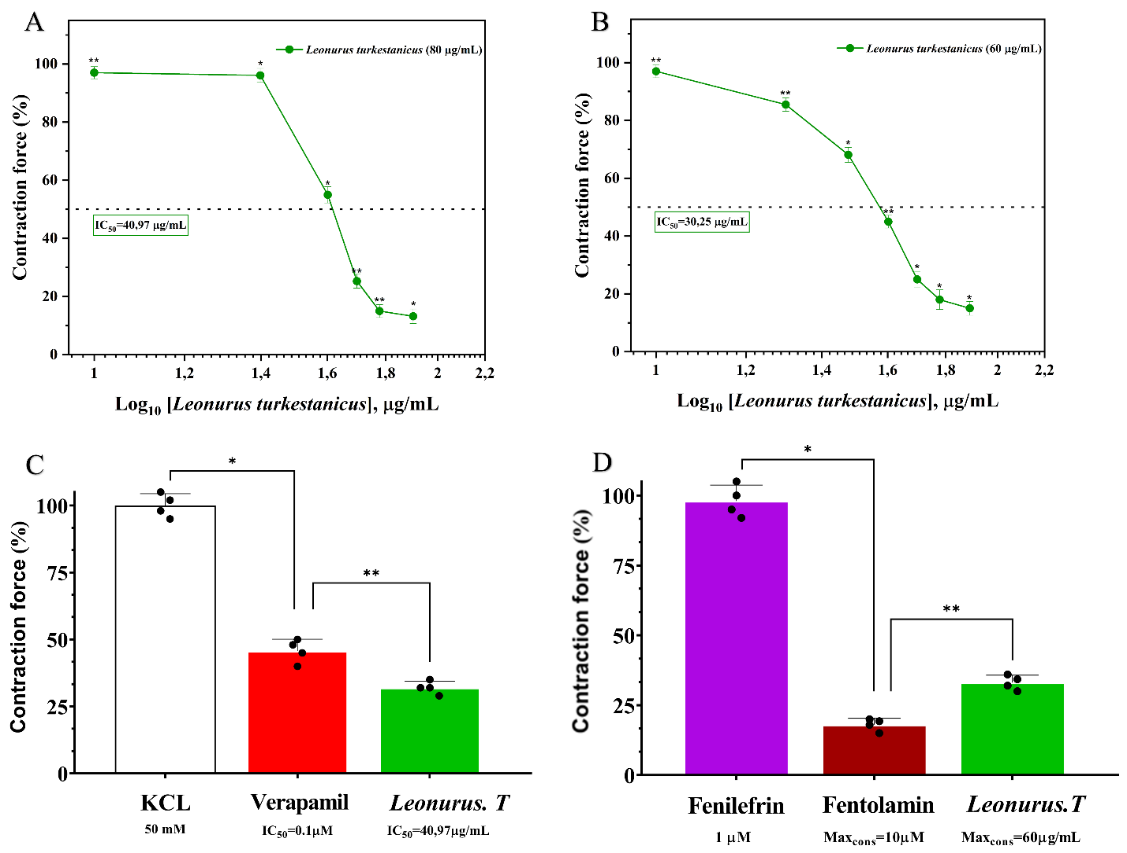
Figure 4 (A) Dose-dependent, linear vasorelaxant effect of extract L. turkestanicus on KCl-induced contraction. (B) Comparison of L. turkestanicus with the calcium channel blocker verapamil. (C) Effect of L. turkestanicus on receptor-operated Ca²⁺ ion channels. (D) Confirmation of F45’s mechanism using the α-adrenergic antagonist phentolamine. Data represent mean ± SEM, n = 3 - 4, p < 0.05.
It is well recognized that vascular smooth muscle contraction is governed not only by voltage-gated L-type Ca²⁺ channels but also by intracellular calcium signaling mechanisms, particularly those associated with the sarcoplasmic reticulum (SR). Both internal calcium release and receptor-operated calcium channels (ROCCs) are essential for maintaining intracellular Ca²⁺ homeostasis and vascular tone [21]. To evaluate whether L. turkestanicus influences receptor-mediated calcium signaling, its effect on phenylephrine (1 μM)-induced contractions in isolated rat aortic rings was assessed. Phenylephrine, an α-adrenoceptor agonist, induces vasoconstriction predominantly through calcium release from the SR and the activation of ROCCs at the plasma membrane. Our experiments revealed that L. turkestanicus markedly attenuated phenylephrine-evoked contractions, exhibiting strong vasorelaxant properties. At the highest tested concentration (60 µg/mL), the extract reduced contractile responses by 85.0 ± 2.4% relative to control (Figure 4(B)). These results suggest that L. turkestanicus limits receptor-operated calcium influx, thereby decreasing intracellular Ca²⁺ levels and suppressing smooth muscle contraction [22]. To further probe this mechanism, we compared its action with phentolamine, a selective α-adrenoceptor antagonist, and with flavonoids known to modulate receptor-mediated calcium signaling. Phentolamine (10 μM) alone inhibited phenylephrine-induced contraction by 85.0 ± 2.6%. Interestingly, co-application of L. turkestanicus (60 µg/mL) with phentolamine produced an additional 33.07 ± 2.7% relaxation (Figure 4(D)), indicating additive or potentially synergistic effects. These findings support the hypothesis that L. turkestanicus interferes with α-adrenoceptor-mediated calcium signaling, most likely by blocking ROCCs and potentially by inhibiting subsequent mobilization of SR calcium stores. Thus, the vasorelaxant activity of L. turkestanicus appears to involve a dual mechanism—both inhibition of L-type Ca²⁺ channels and modulation of receptor-operated calcium entry—highlighting its therapeutic potential in conditions of vascular dysfunction and hypertension [23].
A study on how endothelial mechanisms mediate the relaxant response elicited by L. turkestanicus
The vascular endothelium is a key regulator of vascular tone, largely through the release of local mediators such as nitric oxide (NO), which represents the primary endothelium-derived vasodilator. Structural or functional impairment of the endothelium, known as endothelial dysfunction (ED), is a major contributor to the pathogenesis of cardiovascular diseases, including hypertension and atherosclerosis [24,25].
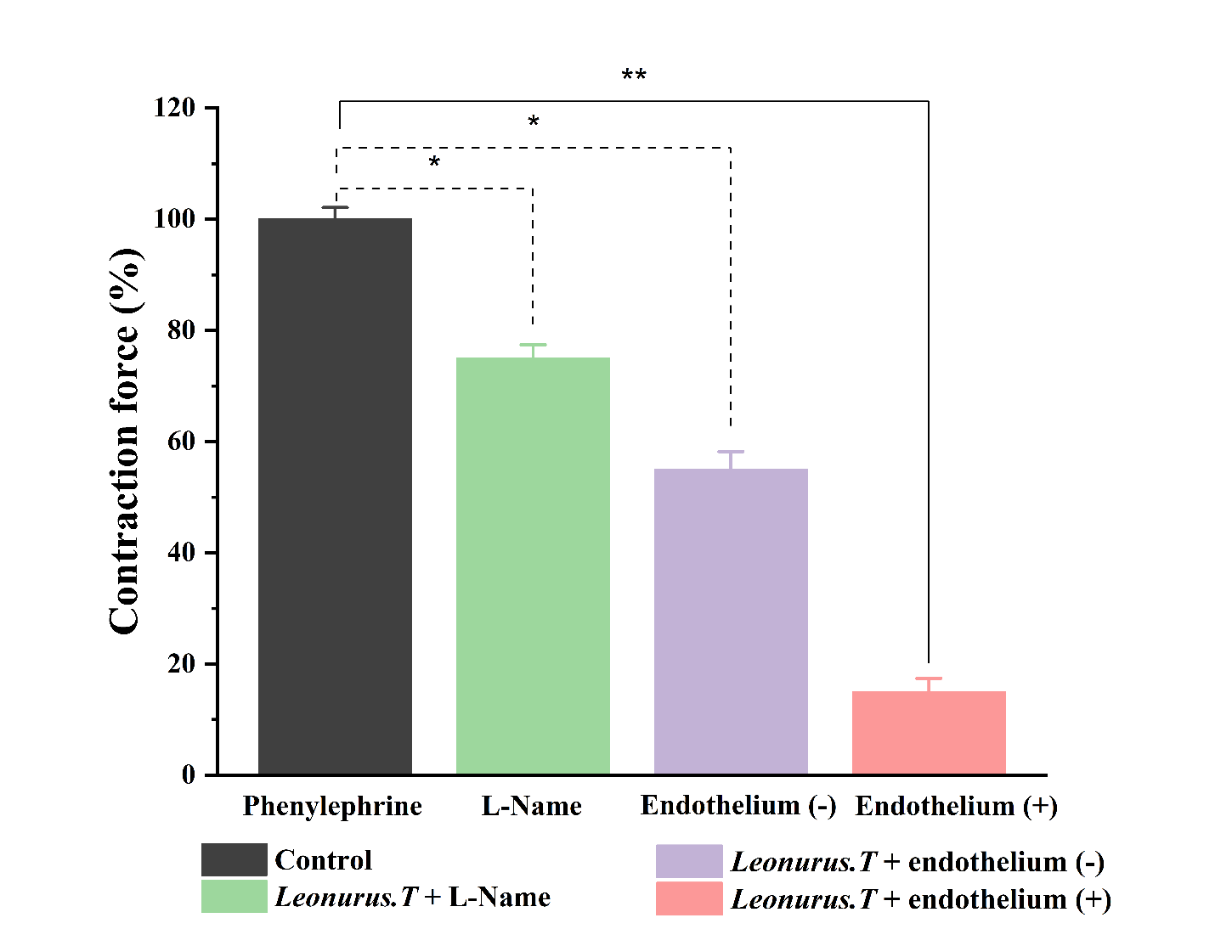
Figure 5 Relaxant effect of L. turkestanicus on the contraction induced by Phe 1 μM in the undenuded and denuded of the rat aortic blood vessel endothelial layer. Contraction force elicited by 1 μM Phe was taken as 100% of control. (in all cases *p < 0.05, **p < 0.01; n = 5).
ED typically develops due to an imbalance between vasodilatory and vasoconstrictive factors, often exacerbated by oxidative stress and modifiable risk factors such as smoking, poor nutrition, and metabolic disorders [22]. Among endothelial mediators, NO plays a central role; it is synthesized from L-arginine by endothelial nitric oxide synthase (eNOS), whose activation is calcium–calmodulin dependent and negatively modulated by its binding to caveolin. Physiological stimuli such as acetylcholine or bradykinin promote dissociation of eNOS from caveolin, thereby enhancing NO release. Additionally, statins have been reported to improve endothelial function by disrupting the eNOS–caveolin complex and increasing NO bioavailability [26]. Conversely, oxidative stress reduces NO signaling by enhancing reactive oxygen species (ROS) production, which rapidly inactivates NO and diminishes its vasodilatory capacity. Under physiological conditions, NO triggers smooth muscle relaxation through the NO/cGMP/PKG pathway, leading to reduced intracellular Ca²⁺ levels and vasodilation [27]. To assess the role of the endothelium in the vasorelaxant response to L. turkestanicus, experiments were performed on both endothelium-intact and endothelium-denuded rat aortic rings. Compared to the control (100% contraction), endothelium removal significantly attenuated the relaxant effect of L. turkestanicus, with contractions maintained at 55.0 ± 3.2% of control. To further validate the involvement of NO signaling, additional assays were performed in the presence of L-NAME, a non-selective nitric oxide synthase (NOS) inhibitor. Under these conditions, vascular relaxation was reduced to 75.0 ± 2.4% relative to endothelium-intact responses (Figure 5).
Molecular docking studies
During our in silico experiments, we investigated the interactions between several flavonoids (Table 2) (apigenin, catechin, ferulic acid, gallic acid, hyperoside, isoquercetin, isorhamnetin, p-coumaric acid, quercetin, and rutin) and different calcium-related channels and transporters, including Ca²⁺-ATPase, L-type calcium channels, R-type calcium channels, NCX1 (sodium-calcium exchanger), RyR1 (ryanodine receptor type 1), and SERCA (sarcoplasmic/endoplasmic reticulum calcium ATPase). The study focused on analyzing the binding affinities, the types of bonds formed during the interactions, and the resulting binding energies. These findings allowed us to better understand how flavonoids may modulate calcium signaling by stabilizing or inhibiting the activity of these channels and transporters through specific molecular interactions.
Table 2 Two-dimensional (2D) and three-dimensional (3D) molecular structures of selected flavonoids.
Flavonoids |
2D |
3D |
Apigenin |
|
|
Catechin |
|
|
Ferulic acid |
|
|
Gallic acid |
|
|
Hyperoside |
|
|
Isoquercetin |
|
|
Isorhamentin |
|
|
P-coumaric acid |
|
|
Quercetin |
|
|
Rutin |
|
|
Ca2+ L type channel
L-type calcium channels (LTCCs) are a subclass of voltage-gated calcium channels that mediate a sustained influx of Ca²⁺ ions in response to membrane depolarization. They are widely distributed throughout the body and play an essential role in diverse physiological processes. In the cardiovascular system, LTCCs are crucial for smooth muscle contraction, vascular tone regulation, and cardiac excitation–contraction coupling. In neurons, they contribute to neurotransmitter release and synaptic plasticity, while in endocrine tissues they regulate hormone secretion. Thus, LTCCs are central to maintaining cellular signaling and overall calcium homeostasis. In smooth muscle, LTCCs serve as the primary pathway for Ca²⁺ entry, directly controlling contraction and relaxation mechanisms that underlie vascular function and blood pressure regulation. Dysregulation of these channels has been linked to hypertension, ischemia, arrhythmias, and neurodegenerative disorders. Given their broad physiological significance, LTCCs represent an important pharmacological target. Recent interest has grown in natural compounds, particularly flavonoids, due to their potential to modulate LTCC activity. By influencing calcium influx and intracellular signaling pathways, flavonoids may provide novel therapeutic avenues for treating cardiovascular and neurological diseases.
Table 3 Molecular interaction of flavonoids with the Ca²⁺ L-type ion channel: Binding energy and Ki values.
Flovanoids |
Affinity (kkal/mol) |
Ki value |
Apigenin |
‒8.6 |
0.5 µM |
Catechin |
‒9.0 |
0.25 µM |
Ferulic |
‒6.3 |
24 µM |
Gallic acid |
‒6.1 |
33.5 µM |
Hyperoside |
‒9.7 |
0.076 µM |
Isoquersetin |
‒9.2 |
0.2 µM |
Isorahmetin |
‒7.5 |
3.17 µM |
P-coumaric acid |
‒6.0 |
39.7 µM |
Quercetin |
‒8.8 |
0.35 µM |
Rutin |
‒9.3 |
0.15 µM |
Among the investigated flavonoids, hyperoside demonstrated one of the strongest inhibitory effects on the Ca²⁺ L-type channel. Docking results showed that it bound tightly to the active site at the coordinates center_x = 168.279698, center_y = 176.439484 and center_z = 180.397000, with a minimum binding energy corresponding to an inhibition constant of Ki = 0.076 µM (Table 3). Such a low Ki value reflects very high binding affinity, suggesting that hyperoside acts as a potent and stable inhibitor of the channel.
Detailed binding analysis revealed multiple interactions with key amino acid residues within the channel. Hyperoside formed conventional hydrogen bonds with GLU A:591, TYR A:1035, ASN A:596, ASP A:598 and TYR A:585, which are important for stabilizing the polar environment of the pore and maintaining channel conformation. Hydrophobic stabilization was provided by π–alkyl interactions with VAL A:592 and LEU F:269, while ASP A:598 also participated in a carbon–hydrogen bond. Additional non-covalent contributions included van der Waals interactions with ASP A:586 and an amide–π stacked interaction with TYR A:585, which strengthened aromatic stabilization. An unfavorable donor–donor interaction was noted with ASN A:596, reflecting steric and electronic constraints of the binding pocket (Figure 6).
These diverse interactions indicate that hyperoside simultaneously engages polar, charged, and hydrophobic residues that are essential for the structural stability and gating function of the Ca²⁺ L-type channel. Such a broad interaction profile likely contributes to its high inhibitory potential by restricting the conformational flexibility of the channel and reducing calcium ion conductance.
Since Ca²⁺ L-type channels play a central role in excitation–contraction coupling in cardiac and vascular smooth muscle cells, as well as in regulating neuronal activity, their inhibition by flavonoids such as hyperoside may have significant pharmacological implications. By stabilizing the channel in a less conductive state, hyperoside could help attenuate abnormal calcium influx, offering potential therapeutic benefits in conditions such as hypertension, cardiac hypertrophy, arrhythmias, and ischemia-induced injury.
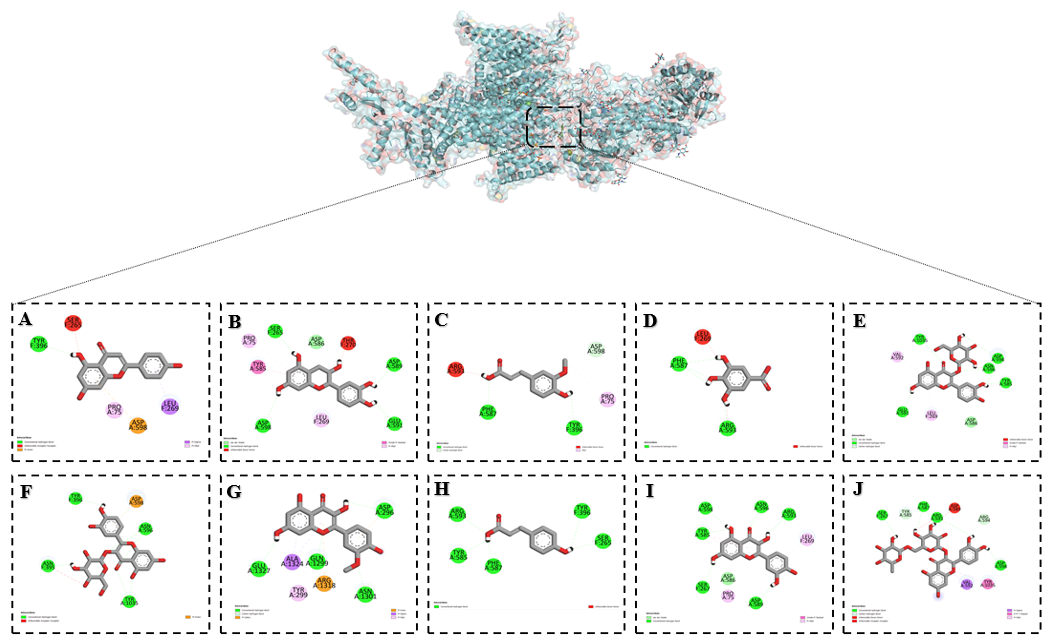
Figure 6 Binding interactions of flavonoids with the Ca²⁺ L-type channel as predicted by molecular docking: (A) Apigenin, (B) Catechin, (C) Ferulic acid, (D) Gallic acid, (E) Hyperoside, (F) Isoquercetin, (G) Isorhamentin, (H) p-Coumaric acid, (I) Quercetin and (J) Rutin.
Ca²⁺-ATPases
Calcium ions (Ca²⁺) are essential second messengers that regulate a wide range of cellular processes, including muscle contraction, neurotransmitter release, gene expression, and cell survival. Maintaining precise intracellular Ca²⁺ homeostasis is therefore critical for normal physiological function. Among the key regulators of calcium dynamics are the P-type Ca²⁺-ATPases, a family of ATP-driven ion pumps responsible for actively transporting Ca²⁺ against its electrochemical gradient. The most well-characterized members include the plasma membrane Ca²⁺-ATPase (PMCA) and the sarco/endoplasmic reticulum Ca²⁺-ATPase (SERCA). PMCA functions to extrude Ca²⁺ from the cytoplasm into the extracellular space, while SERCA pumps Ca²⁺ into the sarcoplasmic or endoplasmic reticulum, thereby replenishing intracellular stores and maintaining low cytosolic Ca²⁺ concentrations. These transporters utilize the energy derived from ATP hydrolysis to drive conformational changes that allow high-affinity binding and translocation of Ca²⁺ ions across membranes.
Dysfunction of Ca²⁺-ATPases has been associated with a variety of pathological conditions, including cardiovascular diseases, diabetes, neurodegenerative disorders, and cancer. In cardiomyocytes, impaired SERCA activity contributes to abnormal Ca²⁺ handling, leading to contractile dysfunction and heart failure. Similarly, aberrant PMCA function has been linked to altered neuronal excitability and neurodegeneration. As a result, Ca²⁺-ATPases have emerged as critical therapeutic targets, and considerable research is focused on identifying natural or synthetic modulators that can restore or enhance their activity. Given the central role of Ca²⁺-ATPases in calcium signaling and cellular homeostasis, investigating their interaction with bioactive compounds provides valuable insights into potential pharmacological strategies aimed at modulating calcium-dependent physiological and pathological processes.
Table 4 Molecular interaction of flavonoids with the Ca²⁺-ATPase: Binding energy and Ki Values.
Flovanoids |
Affinity (kkal/mol) |
Ki value |
Apigenin |
‒8.9 |
0.3 µM |
Catechin |
‒8.7 |
0.5 µM |
Ferulic |
‒6.6 |
14.4 µM |
Gallic acid |
‒6.0 |
37 µM |
Hyperoside |
‒10.4 |
0.02 µM |
Isoquersetin |
‒10.0 |
0.04 µM |
Isorahmetin |
‒9.1 |
0.21 µM |
P-coumaric acid |
‒6.4 |
20.2 µM |
Quercetin |
‒8.9 |
0.3 µM |
Rutine |
‒11.0 |
0.008 µM |
The results clearly demonstrate that among the studied flavonoids, rutin exhibited the most stable binding and the strongest inhibitory activity (Table 4). This trend of inhibitory potency followed the order: Rutin > Hyperoside > Isoquercetin > Isorhamnetin > Apigenin ≈ Quercetin > Catechin > Ferulic acid > p-Coumaric acid > Gallic acid, highlighting rutin as the most effective inhibitor within this group. Docking analysis revealed that rutin interacted with several key residues within the Ca²⁺-ATPase active site. It formed conventional hydrogen bonds with Thr A:353, Gly A:625, and Arg A:559, which contribute to stabilizing the protein conformation and ligand anchoring. A carbon–hydrogen bond was observed with Glu A:439, further reinforcing the binding orientation. Unfavorable donor–donor and acceptor–acceptor interactions were detected with Asn A:359 and Asp A:702, reflecting the electrostatic constraints of the binding pocket. In addition, Met A:494 participated in a π–sulfur interaction, while Phe A:487 engaged in a π–π stacking interaction, which are important for stabilizing aromatic and hydrophobic contacts. Hydrophobic stabilization was further supported by π–alkyl interactions with Ala A:516 and Leu A:561, whereas Arg A:559 also contributed to π–cation interactions, providing additional electrostatic stabilization (Figure 7). These multiple interactions indicate that rutin binds selectively and strongly to Ca²⁺-ATPase, targeting residues critical for its structural stability and catalytic activity, thereby providing a plausible molecular basis for its potent inhibitory effect.
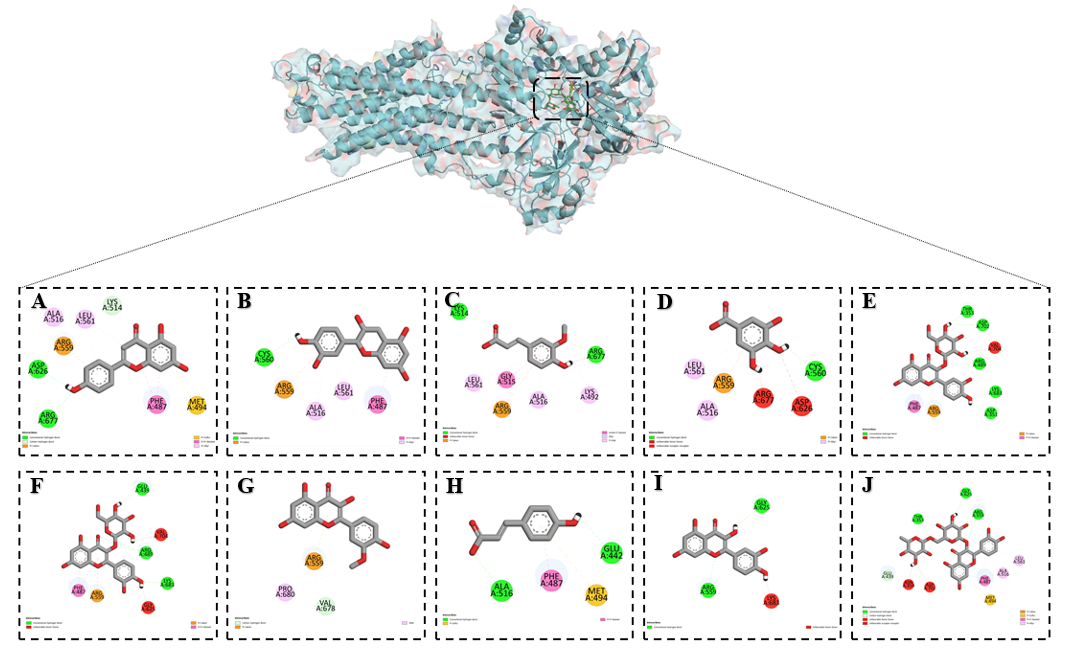
Figure 7 Binding interactions of flavonoids with the Ca²⁺-ATPase as predicted by molecular docking: (A) Apigenin, (B) Catechin, (C) Ferulic acid, (D) Gallic acid, (E) Hyperoside, (F) Isoquercetin, (G) Isorhamentin, (H) p-Coumaric acid, (I) Quercetin and (J) Rutin.
Ca²⁺ R-type channel
R-type calcium channels (CaV2.3) belong to the family of voltage-gated calcium channels that regulate the influx of Ca²⁺ ions across the plasma membrane in response to changes in membrane potential. Unlike the well-characterized L-type, N-type, and T-type channels, R-type channels display unique electrophysiological and pharmacological properties. They are activated at relatively high voltages, exhibit intermediate kinetics of activation and inactivation, and contribute to a wide range of physiological processes. R-type channels are widely expressed in the central nervous system, particularly in the cerebral cortex, hippocampus, and cerebellum, where they are involved in synaptic transmission, dendritic calcium signaling, and neuronal excitability. They also play important roles in regulating gene expression, neurotransmitter release, and synaptic plasticity. In addition to the nervous system, R-type calcium channels have been identified in cardiac and smooth muscle tissues, where they may influence vascular tone and contribute to excitation–contraction coupling. Dysregulation of CaV2.3 channels has been linked to several pathological conditions, including epilepsy, neuropathic pain, ischemia, and cardiovascular dysfunction. Although selective pharmacological blockers of R-type channels are limited, natural products and synthetic compounds with modulatory activity are being increasingly studied for their therapeutic potential. Given their broad physiological roles and association with disease, R-type calcium channels represent an important molecular target for understanding calcium-dependent signaling mechanisms and for the development of novel therapeutic agents.
In our in silico study, the interaction of flavonoids with the Ca²⁺ R-type channel revealed a minimum binding energy of –8.7 kcal/mol (Table 5). The inhibitory potency of the tested flavonoids can be arranged in the following order: Quercetin > Isorhamnetin > Apigenin ≈ Rutin > Hyperoside > Isoquercetin ≈ Catechin > p-Coumaric acid > Ferulic acid > Gallic acid.
Table 5 Molecular interaction of flavonoids with the Ca²⁺ R-type channel: Binding energy and Ki Values.
Flovanoids |
Affinity (kkal/mol) |
Ki value |
Apigenin |
‒8.5 |
0.96 µM |
Catechin |
‒7.9 |
1.6 µM |
Ferulic |
‒6.0 |
39.66 µM |
Gallic acid |
‒5.5 |
92.3 µM |
Hyperoside |
‒8.0 |
1.35 µM |
Isoquersetin |
‒7.9 |
1,6 µM |
Isorahmetin |
‒8.4 |
0.68 µM |
P-coumaric acid |
‒6.5 |
17.04 µM |
Quercetin |
‒8.7 |
0.4 µM |
Rutine |
‒8.2 |
0.94 µM |
Among the studied flavonoids, quercetin demonstrated the strongest inhibitory effect on the Ca²⁺ R-type channel. Docking analysis revealed that quercetin established multiple stabilizing interactions with key residues in the active site. It formed conventional hydrogen bonds with ASP A:1465, GLN A:117, ARG A:1472, HIS A:1480, THR A:1342, and ASN A:1466, ensuring strong polar interactions and ligand anchoring. In addition, LYS A:1345 participated in a π–cation interaction, while TYR A:1469 engaged in a π–π T-shaped interaction, both of which contributed to enhanced electrostatic and aromatic stabilization. Furthermore, PRO A:1479 was involved in π–alkyl interactions, reinforcing hydrophobic stabilization within the binding pocket (Figure 8).
These diverse interactions indicate that quercetin targets residues critical for the conformational stability and ion transport function of the Ca²⁺ R-type channel, thereby providing a molecular basis for its potent inhibitory activity.
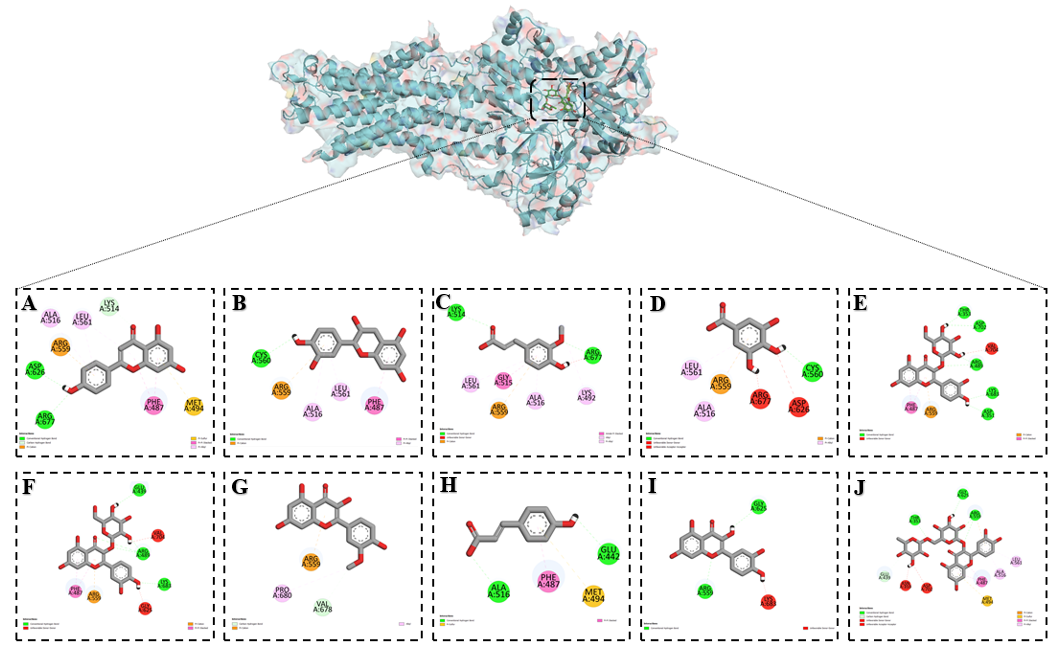
Figure 8 Binding interactions of flavonoids with the Ca²⁺ R-type channel as predicted by molecular docking: (A) Apigenin, (B) Catechin, (C) Ferulic acid, (D) Gallic acid, (E) Hyperoside, (F) Isoquercetin, (G) Isorhamentin, (H) p-Coumaric acid, (I) Quercetin and (J) Rutin.
NCX1
The sodium–calcium exchanger 1 (NCX1) is a membrane transport protein that plays a central role in maintaining intracellular calcium homeostasis. It operates by coupling the movement of sodium (Na⁺) and calcium (Ca²⁺) ions across the plasma membrane in opposite directions, typically extruding one Ca²⁺ ion in exchange for the influx of three Na⁺ ions. This electrogenic process is essential for the rapid removal of cytosolic Ca²⁺ following cellular excitation. NCX1 is widely expressed in excitable tissues such as the heart, brain, and skeletal muscle, as well as in smooth muscle and various non-excitable cells. In cardiomyocytes, NCX1 is particularly important for diastolic relaxation, as it facilitates the clearance of Ca²⁺ after contraction. By balancing intracellular calcium, NCX1 contributes to excitation–contraction coupling, membrane potential regulation, and overall cellular signaling.
Dysregulation of NCX1 activity has been associated with several pathological conditions. In the heart, altered NCX1 function contributes to arrhythmias, ischemia–reperfusion injury, and heart failure. The results of our molecular docking analysis demonstrated that the interaction of flavonoids with the NCX1 channel yielded a minimum binding energy of – 9.1 kcal/mol (Table 5). Based on the binding affinities, the inhibitory potency of the flavonoids can be ranked as follows: Apigenin = Hyperoside = Rutin > Quercetin > Isoquercetin = Isorhamnetin > Catechin > p-Coumaric acid > Gallic acid > Ferulic acid. This ranking highlights apigenin, hyperoside, and rutin as the strongest inhibitors among the tested compounds.
Table 6 Molecular interaction of flavonoids with the Ca²⁺ R-type channel: Binding energy and Ki Values.
Flovanoids |
Affinity (kkal/mol) |
Ki value |
Apigenin |
‒ 9.1 |
0.21 µM |
Catechin |
‒ 8.8 |
0.35 µM |
Ferulic |
‒ 5.8 |
55.6 µM |
Gallic acid |
‒ 6.1 |
33.5 µM |
Hyperoside |
‒ 9.1 |
0.21 µM |
Isoquersetin |
‒ 8.9 |
0.29 µM |
Isorahmetin |
‒ 8.9 |
0.29 µM |
P-coumaric acid |
‒ 6.6 |
14.4 µM |
Quercetin |
‒ 9.0 |
0.25 µM |
Rutine |
‒ 9.1 |
0.21 µM |
Among the studied flavonoids, apigenin, hyperoside, and rutin exhibited the most pronounced inhibitory effects on the NCX1 (Na⁺/Ca²⁺ exchanger 1) channel. Molecular docking analysis demonstrated that these compounds established a wide range of stabilizing interactions within the binding site of the exchanger, reflecting both polar and hydrophobic complementarity. Conventional hydrogen bonds were formed with GLU A:244, THR A:172, THR A:760, ARG A:63, SER A:66, ASP A:62, GLN A:712, and GLY A:238, residues that play key roles in maintaining the structural integrity of the exchanger and in coordinating ion binding during transport. Hydrogen bonding with these residues suggests that the flavonoids stabilize the protein in a conformation less favorable for efficient ion exchange, thereby reducing its activity (Figure 3). Additional stabilizing forces were provided by π–anion interactions with ASP A:829 and ARG A:63, which are charged residues involved in the electrostatic regulation of ion translocation. Hydrophobic contacts through π–alkyl interactions were observed with ALA A:830, LEU A:222, LEU A:102, and LYS A:751, reinforcing ligand accommodation within the hydrophobic core of the channel. Furthermore, VAL A:99 participated in a π–σ interaction, while GLU A:715 and ARG A:63 contributed to π–cation interactions, which are essential for stabilizing charged and aromatic regions of the ligand within the active pocket. Functionally, NCX1 operates bidirectionally, extruding Ca²⁺ in exchange for Na⁺ entry (forward mode) under physiological conditions, or importing Ca²⁺ in exchange for Na⁺ efflux (reverse mode) during pathological states such as ischemia and calcium overload. The ability of apigenin, hyperoside, and rutin to engage residues critical for ion coordination and conformational gating suggests that these flavonoids may impair both forward and reverse exchange modes, thereby attenuating excessive calcium influx.
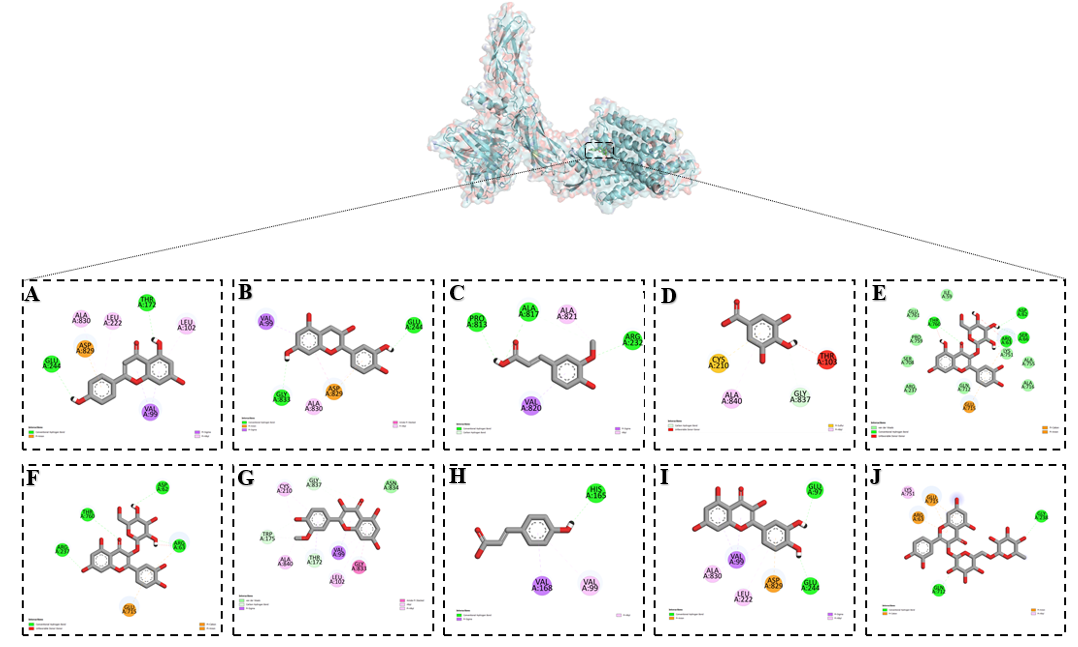
Figure 4 Binding interactions of flavonoids with the NCX1 channel as predicted by molecular docking: (A) Apigenin, (B) Catechin, (C) Ferulic acid, (D) Gallic acid, (E) Hyperoside, (F) Isoquercetin, (G) Isorhamentin, (H) p-Coumaric acid, (I) Quercetin and (J) Rutin.
RyR2
The ryanodine receptor type 2 (RyR2) is a large intracellular calcium release channel located in the membrane of the sarcoplasmic reticulum (SR) in cardiac muscle cells. As a member of the ryanodine receptor family, RyR2 plays a central role in excitation–contraction coupling by mediating the release of stored Ca²⁺ from the SR into the cytoplasm during the action potential. This rapid increase in intracellular calcium concentration triggers contraction of cardiomyocytes. RyR2 is activated by Ca²⁺ influx through L-type calcium channels in the plasma membrane, a mechanism known as calcium-induced calcium release (CICR). This process ensures a finely tuned amplification of calcium signals, which is essential for normal cardiac rhythm and contractility. Dysfunction of RyR2 has been implicated in a range of pathological conditions. Mutations or abnormal regulation of this receptor can lead to catecholaminergic polymorphic ventricular tachycardia (CPVT), arrhythmias, heart failure, and sudden cardiac death. Excessive Ca²⁺ leak from RyR2 during diastole contributes to impaired contractility and arrhythmogenic triggers in diseased hearts. Because of this, RyR2 is considered an important therapeutic target, and both natural and synthetic compounds are under investigation for their potential to stabilize RyR2 function and reduce pathological calcium leak. Given its crucial role in regulating intracellular calcium dynamics and maintaining cardiac rhythm, RyR2 remains a key protein of interest in cardiovascular physiology and pharmacology, as well as in the search for novel therapeutic agents to treat calcium-related cardiac disorders. The results of our in silico analysis demonstrated that flavonoids interact with the RyR2 channel with varying degrees of affinity. Among the tested compounds, isoquercetin, isorhamnetin, and quercetin displayed the strongest binding capacity. These flavonoids were localized to the active site of the channel with coordinates center_x = 9.181367, center_y = 17.019000 and center_z = 27.203000, where they established stable interactions. The lowest binding energy observed was –7.9 kcal/mol, indicating high binding affinity and strong inhibitory potential (Table 7). The ability of these flavonoids to achieve such stable binding suggests that they may significantly influence the functional state of RyR2. Since RyR2 mediates calcium-induced calcium release (CICR) from the sarcoplasmic reticulum, its overactivation or destabilization is closely associated with pathological calcium leak, arrhythmogenesis, and impaired cardiac contractility.
Table 7 molecular interaction of flavonoids with the Ca²⁺ R-type channel: Binding energy and Ki Values.
Flovanoids |
Affinity (kkal/mol) |
Ki value |
Apigenin |
‒7.5 |
3.1 µM |
Catechin |
‒7.3 |
4.4 µM |
Ferulic |
‒5.9 |
46.9 µM |
Gallic acid |
‒6.5 |
17 µM |
Hyperoside |
‒7.5 |
3.1 µM |
Isoquersetin |
‒7.9 |
1.6 µM |
Isorahmetin |
‒7.9 |
1.6 µM |
P-coumaric acid |
‒6.4 |
20.2 µM |
Quercetin |
‒7.9 |
1.6 µM |
Rutine |
‒7.8 |
1.9 µM |
Docking analysis showed that isoquercetin, isorhamnetin and quercetin strongly bound to the RyR2 channel and exerted inhibitory effects. These flavonoids interacted with amino acid residues PRO B:707, GLU A:701, THR A:700, GLU A:711, GLU B:711, GLY A:786, LEU A:787 and GLY B:710. Proline (PRO B:707) contributes to structural flexibility of the loop region. Glutamic acid residues (GLU A:701, GLU A:711, GLU B:711) are negatively charged and play a role in electrostatic stabilization and ion coordination. Threonine (THR A:700) is involved in polar interactions and stabilizes hydrogen bonding within the pore. Glycine residues (GLY A:786 and GLY B:710) provide conformational flexibility and allow local structural adjustments. Leucine (LEU A:787) is hydrophobic and contributes to stabilizing the transmembrane environment (Figure 9). Through these interactions the flavonoids anchor within residues that are critical for conformational stability and ion release function of RyR2, which explains their strong inhibitory effects on calcium-induced calcium release.
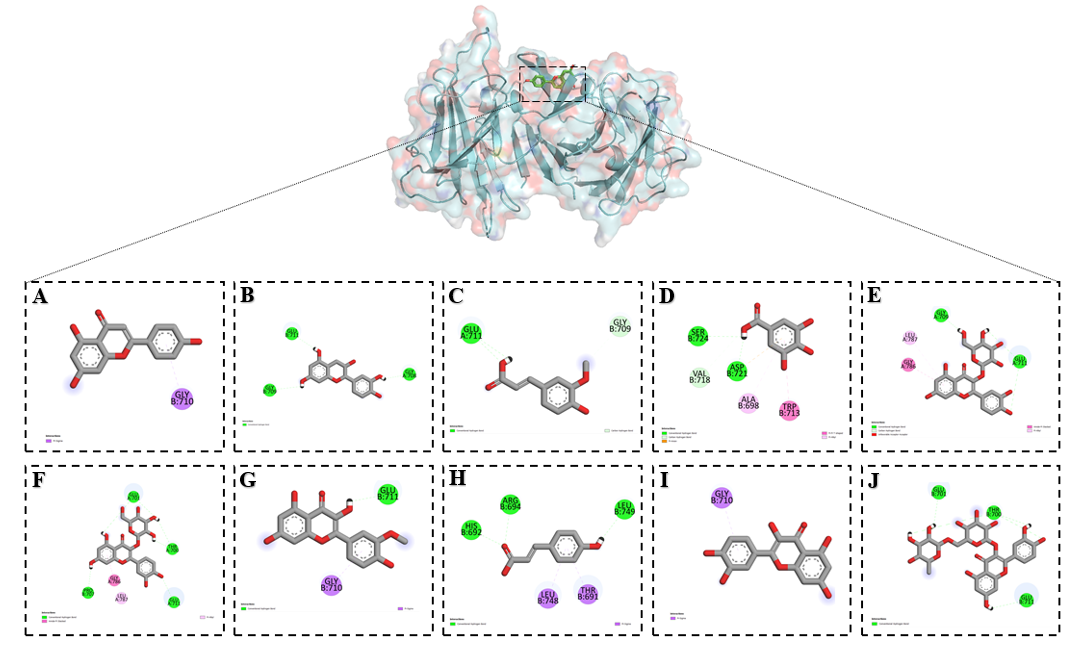
Figure 9 Binding interactions of flavonoids with the NCX1 channel as predicted by molecular docking: (A) Apigenin, (B) Catechin, (C) Ferulic acid, (D) Gallic acid, (E) Hyperoside, (F) Isoquercetin, (G) Isorhamentin, (H) p-Coumaric acid, (I) Quercetin and (J) Rutin.
SERCA
The sarco/endoplasmic reticulum Ca²⁺-ATPase (SERCA) is a membrane-bound P-type ATPase that plays a central role in maintaining intracellular calcium homeostasis. It is located in the membrane of the sarcoplasmic and endoplasmic reticulum, where it actively pumps Ca²⁺ ions from the cytosol back into the lumen of these organelles using the energy derived from ATP hydrolysis. This process is essential for lowering cytosolic calcium concentration after stimulation and for replenishing intracellular Ca²⁺ stores.
In muscle cells, SERCA is a key component of excitation–contraction coupling. During relaxation, SERCA rapidly removes Ca²⁺ from the cytoplasm and transports it into the sarcoplasmic reticulum, allowing muscle fibers to return to a resting state. Several isoforms of SERCA exist (SERCA1, SERCA2, SERCA3), each with tissue-specific expression and physiological roles. SERCA2a, in particular, is highly expressed in cardiomyocytes and is essential for normal cardiac contractility. Dysfunction of SERCA activity has been linked to various diseases. In the cardiovascular system, reduced SERCA2a function contributes to impaired calcium cycling, diastolic dysfunction, and the progression of heart failure. In non-muscle tissues, altered SERCA activity is associated with metabolic disorders, neurodegeneration, and cancer. Because of its fundamental role, SERCA is considered a critical therapeutic target, and both pharmacological agents and gene therapy approaches (such as SERCA2a gene transfer) are under investigation to restore or enhance its function. Given its importance in calcium handling, energy metabolism, and cellular signaling, SERCA remains a focus of biomedical research, particularly in the context of cardiovascular diseases, where strategies aimed at improving SERCA activity hold promise for novel therapeutic interventions.
Based on the results of our in silico experiments, the interaction of flavonoids with the SERCA channel revealed a minimum binding energy of – 9.6 kcal/mol. This lowest energy value was observed for rutin, indicating its strong and stable binding within the active site of the channel. The binding pocket was defined at the coordinates center_x = 37.179317, center_y = 34.354870, and center_z = 58.830812, where rutin established multiple stabilizing interactions with critical amino acid residues (Table 8). These findings suggest that rutin possesses the highest inhibitory potential among the tested flavonoids. Its ability to bind at such a low energy state highlights a strong affinity toward SERCA, which may significantly influence calcium transport by reducing pump activity. This provides a plausible molecular basis for the role of rutin as a potent modulator of intracellular calcium homeostasis through SERCA inhibition.
Table 8 Molecular interaction of flavonoids with the Ca²⁺ R-type channel: Binding energy and Ki Values.
Flovanoids |
Affinity (kkal/mol) |
Ki value |
Apigenin |
‒8.0 |
1.35 µM |
Catechin |
‒8.0 |
1.35 µM |
Ferulic |
‒5.9 |
46.9 µM |
Gallic acid |
‒6.0 |
39.7 µM |
Hyperoside |
‒9.1 |
0.21 µM |
Isoquersetin |
‒9.0 |
0.25 µM |
Isorahmetin |
‒8.0 |
1.35 µM |
P-coumaric acid |
‒5.6 |
77.9 µM |
Quercetin |
‒8.4 |
0.67 µM |
Rutine |
‒9.6 |
0.09 µM |
Docking analysis revealed that rutin strongly interacted with the SERCA channel and contributed to its inhibition. The compound formed conventional hydrogen bonds with LYS A:728, ARG A:334, LYS A:120, and MET A:126, anchoring the ligand within the binding pocket. In addition, ALA A:736 established a π–alkyl interaction, while GLU A:125 participated in a π–anion interaction, further stabilizing the complex. An unfavorable donor–donor interaction was observed with ARG A:334, which reflects steric and electrostatic constraints within the active site. These multiple interactions suggest that rutin targets residues essential for the structural stabilization and ion transport function of SERCA, thereby providing a molecular explanation for its inhibitory effect.
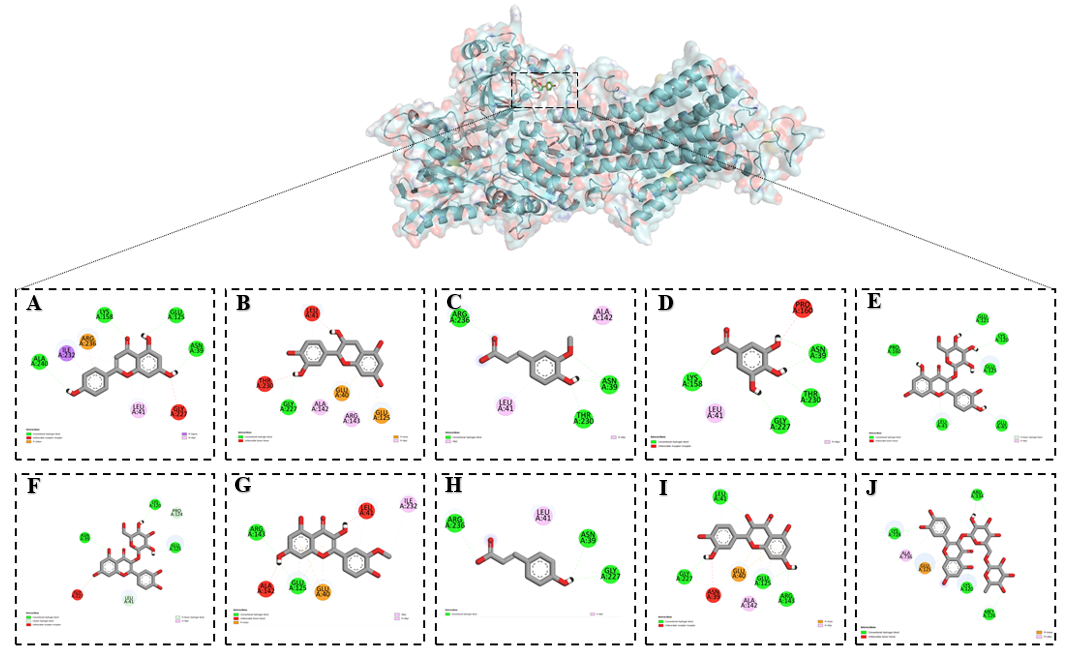
Figure 10 Binding interactions of flavonoids with the NCX1 channel as predicted by molecular docking (A) Apigenin, (B) Catechin, (C) Ferulic acid, (D) Gallic acid, (E) Hyperoside, (F) Isoquercetin, (G) Isorhamentin, (H) p-Coumaric acid, (I) Quercetin and (J) Rutin.
Discussion
This study provides a comprehensive comparative evaluation of the phytochemical composition and calcium-modulating mechanisms underlying the vasorelaxant and antihypertensive activities of the Leonurus turkestanicus aqueous extract. By integrating HPLC-based metabolite profiling, vascular reactivity assays, and in silico docking analysis, we established both the chemical basis and functional mechanisms contributing to the cardiovascular protective effects of the extract.
Chromatographic profiling confirmed that L. turkestanicus is a rich source of flavonoids and phenolic acids, with rutin, isoquercitrin, hyperoside, quercetin, and apigenin identified as major constituents. The abundance of flavonoid glycosides suggests that these compounds are the principal mediators of the extract’s vascular bioactivity. Compared with previous reports on other Leonurus species, such as L. cardiaca and L. sibiricus, the Uzbek endemic L. turkestanicus demonstrates a distinctive phytochemical profile with higher levels of rutin and hyperoside, both known for their potent antioxidant and endothelial-protective properties.
Functionally, the extract exhibited multi-mechanistic vasorelaxant effects in isolated rat aortic rings. The inhibition of KCl-induced contractions indicates suppression of voltage-dependent L-type Ca²⁺ channels, while the additive response with verapamil confirms shared pharmacological targets. The extract also reduced phenylephrine-evoked contractions, implying modulation of receptor-operated Ca²⁺ channels (ROCCs) and attenuation of α₁-adrenoceptor-mediated Ca²⁺ signaling. The observed synergistic interaction with phentolamine supports this dual modulation of both voltage- and receptor-operated calcium entry pathways.
In addition, endothelium-dependent mechanisms significantly contributed to relaxation. The attenuation of vasodilatory responses by L-NAME suggests that nitric oxide (NO) synthesis plays a crucial role in the extract’s action. These findings indicate that L. turkestanicus enhances endothelial function while simultaneously reducing smooth muscle Ca²⁺ influx—two complementary mechanisms that together maintain vascular tone and improve hemodynamic stability.
The molecular docking analyses provided a mechanistic rationale for these physiological effects by identifying probable molecular targets of the key flavonoids. Among all tested ligands, hyperoside displayed the strongest affinity for L-type Ca²⁺ channels, while rutin exhibited selective binding to Ca²⁺-ATPases (SERCA). Quercetin showed the highest affinity for R-type channels, and apigenin and isoquercitrin were predicted to interact strongly with Na⁺/Ca²⁺ exchangers (NCX1) and RyR2 receptors. The distribution of binding energies and interaction residues suggests a complementary, multi-target modulation of intracellular and membrane-bound calcium transport systems.
Comparatively, hyperoside and rutin demonstrated broader target engagement and stronger affinities than other flavonoids, which aligns with their dominant representation in the chromatographic profile and their well-documented cardiovascular benefits. These results imply that L. turkestanicus exerts its pharmacological effects through a synergistic network of polyphenolic compounds, acting collectively to restore Ca²⁺ homeostasis and suppress vasoconstrictive responses.
Taken together, the Leonurus turkestanicus extract demonstrates three convergent mechanisms of action: (1) direct smooth muscle relaxation via Ca²⁺ channel inhibition; (2) endothelium-dependent NO-mediated vasodilation; and (3) multi-target regulation of calcium transport proteins. The integration of these pathways positions L. turkestanicus as a potent natural modulator of vascular tone. The interactions observed with clinically relevant targets such as L-type Ca²⁺ channels and SERCA further support the plant’s potential as a source of novel antihypertensive and cardioprotective agents.
Moreover, the present findings establish a strong mechanistic link between the molecular docking predictions and the physiological outcomes, bridging computational pharmacology with experimental vascular pharmacodynamics. Future work should include electrophysiological and calcium-imaging validation of these targets and comparative studies with isolated flavonoids to delineate their individual contributions within the extract matrix.
Conclusions
These results validate the traditional medicinal use of L. turkestanicus and highlight its therapeutic potential in hypertension and vascular dysfunction. The combination of phytochemical richness and multi-target calcium modulation positions this plant as a promising source of lead compounds for cardiovascular drug development. Future work should focus on in vivo pharmacological validation, pharmacokinetics, and safety assessment to translate these findings into clinical applications.
Declaration of Generative AI in Scientific Writing
Only minimal assistance was used from QuillBot for paraphrasing selected sentences. All scientific content, interpretation, and conclusions were developed independently by the authors.
CRediT author statement
Gulhayoxon Mirzaalimova – Conceptualization, Methodology, Data curation, Writing – Original Draft. Anvar Zaynabiddinov – Formal analysis, Validation, Visualization. Sirojiddin Omonturdiev – Investigation, Laboratory experiments, Data collection. Dolimjon Inomjonov – in vitro. Izzatullo Abdullaev – Writing – Review & Editing, Project administration. Ulugbek Gayibov – Supervision, Methodology, Funding acquisition. Madina Khomirzayeva – Visualization, Graphical abstract preparation, Literature review.
References
[1] S Goorani, S Zangene and JD Imig. Hypertension: A Continuing Public Healthcare Issue. International Journal of Molecular Sciences 2025; 26(1), 123.
[2] X Wu, J Sha, Q Yin, Y Gu and X He. Global burden of hypertensive heart disease and attributable risk factors, 1990 - 2021: Insights from the global burden of disease study. Scientific Reports 2025; 15, 14594.
[3] I Abdullaev, U Gayibov, S Omonturdiev, S Fotima, S Gayibova and T Aripov. Molecular pathways in cardiovascular disease under hypoxia: Mechanisms, biomarkers, and therapeutic target. The Journal of Biomedical Research 2025; 39(3), 254-269.
[4] AA Abdullaev, DR Inamjanov, DS Abduazimova, SZ Omonturdiyev, UG Gayibov, SN Gayibova and TF Aripov. Sílybum Mariánum’s impact on physiological alterations and oxidative stress in diabetic rats. Biomedical and Pharmacology Journal 2024; 17(2), 1291-1300.
[5] AV Mahmudov, OS Abduraimov, SB Erdonov, UG Gayibov and LY Izotova. Bioecological features of Nigella sativa L. in different conditions of Uzbekistan. Plant Science Today 2022; 9(2), 421-426.
[6] OK Khojimatov, VV Pak and RW Bussmann. Leonurus turkestanicus V.I. Krecz. & Kuprian., Leonurus panzerioides Popov – LAMIACEAE. In: OK Khojimatov, Y Gafforov and RW Bussmann (eds). Ethnobiology of Uzbekistan. Springer, Cham, 2023, p. 481-485.
[7] AQQ Azimova, AX Islomov, SA Maulyanov, DG Abdugafurova, LU Mahmudov, IZ Abdullaev, AS Ishmuratova, SQ Siddikova and IR Askarov. Determination of vitamins and pharmacological properties of Vitis vinifera L. plant fruit part (Mixed Varieties) syrup-honey. Biomedical and Pharmacology Journal 2024; 17(4), 2779-2786.
[8] O Gaibullayeva, A Islomov, D Abdugafurova, B Elmurodov, B Mirsalixov, L Mahmudov, I Adullaev, K Baratov, S Omonturdiev and S Sa’dullayeva. Inula helenium l. root extract in sunflower oil: Determination of its content of water-soluble vitamins and immunity-promoting effect. Biomedical and Pharmacology Journal 2024; 17(4), 2729-2737.
[9] A Abdullaev, I Abdullaev, A Bogbekov, U Gayibov, S Omonturdiev, S Gayibova, M Turahodjayev, K Ruziboev and T Aripov. Antioxidant potential of Rhodiola heterodonta extract: Activation of Nrf2 pathway via integrative in vivo and in silico studies. Trends in Sciences 2025; 22(5), 9521.
[10] OS Zoirovich, AI Ziyoyiddin, ID Raxmatillayevich, ML Umarjonovich, ZM Ravshanovna, GS Narimanovna, GU Gapparjanovich and AT Fatikhovich. The effect of Ájuga Turkestánica on the rat aortic smooth muscle ion channels. Biomedical and Pharmacology Journal 2024; 17(2), 1213-1222.
[11] D Inomjonov, I Abdullaev, S Omonturdiev, A Abdullaev, L Maxmudov, M Zaripova, M Abdullayeva, D Abduazimova, S Menglieva, S Gayibova, M Sadbarxon, U Gayibov and T Aripov. In vitro and in vivo studies of Crategus and Inula helenium extracts: Their effects on rat blood pressure. Trends in Sciences 2025; 22(3), 9158.
[12] S Sodiqova, S Kadirova, A Zaynabiddinov, I Abdullaev, L Makhmudov, U Gayibov, M Yuldasheva, M Xolmirzayeva, R Rakhimov, A Mutalibov and H Karimjonov. Channelopathy activity Of A-41(Propyl Ester of Gallic Acid): Experimental and computational study of antihypertensive activity. Trends in Sciences 2025; 22(9), 10496.
[13] R Sayidaliyeva, S Kadirova, A Zaynabiddinov, I Abdullaev, L Makhmudov, U Gayibov, M Yuldasheva, M Kholmirzayeva, R Rakhimov, A Mutalibov and H Karimjonov. A-51 as A natural calcium channel blocker: An integrative study targeting hypertension. Trends in Sciences 2025; 22(11), 10760.
[14] A Khasanov, I Abdullaev, S Kadirova, M Mamajanov, A Zaynabiddinov, S Omonturdiev, L Makhmudov, D Inomjonov, U Gayibov, R Esanov and A Matchanov. N-2 polyphenol targets vascular calcium channels to exert antihypertensive effects: In vitro and in vivo evaluation. Trends in Sciences 2025; 22(12), 10782.
[15] O Trott and AJ Olson. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 2010; 31(2), 455-461.
[16] M Zaripova, I Abdullaev, A Bogbekov, U Gayibov, S Omonturdiev, R Makhmudov, N Ergashev, G Jabbarova, S Gayibova and T Aripov. In vitro and in silico studies of gnaphalium u. extract: Inhibition of α-amylase and α-glucosidase as a potential strategy for metabolic syndrome regulation. Trends in Sciences 2025; 22(8), 10098.
[17] TF Aripov, AE Zaynabiddinov, MA Xolmirzayeva, HS Ruziboev, RN Rakhimov, DS Abduazimova, AA ugli Abdullaev, HM ugli Karimjonov, SN Gayibova and UG Gayibov. Antioxidant and cardioprotective properties of polyphenolic plant extract of Rhus glabra L. Plant Science Today 2024; 11(3), 3442.
[18] TF Aripov and UG Gayibov. Antiradical and antioxidant activity of the preparation “Rutan” from Rhus coriaria L. Journal of Theoretical and Clinical Medicine 2023; 4, 164-170.
[19] MR Zaripova, SN Gayibova, RR Makhmudov, AA Mamadrahimov, NL Vypova, UG Gayibov, SM Miralimova and TF Aripov. Characterization of Rhodiola heterodonta (Crassulaceae): Phytocomposition, antioxidant and antihyperglycemic activities. Preventitive Nutrition Food Science 2024; 29(2), 135-145.
[20] AV Mahmudov, OS Abduraimov, SB Erdonov, AL Allamurotov, OT Mamatkasimov, UG Gayibov and LY Izotova. Seed productivity of Linum usitatissimum L. in different ecological conditions of Uzbekistan. Plant Science Today 2022; 9(4), 1090-1101.
[21] A Sandoo, JJCSV van Zanten, GS Metsios, D Carroll and GD Kitas. The endothelium and its role in regulating vascular tone. The Open Cardiovascular Medicine Journal 2010; 23(4), 302-312.
[22] TF Aripov, UG Gayibov, SN Gayibova, AA ugli Abdullaev, DS kizi Abduazimova, YI Oshchepkova and SI Salikhov. In vitro antioxidant and antiradical activity of the total polyphenols (substances of the antiviral drug Rutan) of the leaves of tannic sumach Rhus coriaria L. Khimiya Rastitel’nogo Syr’ya. 2024; (4), 138-148.
[23] UG Gayibov, EJ Komilov, RN Rakhimov, NA Ergashev, NG Abdullajanova, MI Asrorov and TF Aripov. Influence of new polyphenol compound from Euphorbia plant on mitochondrial function. Journal of Microbiology Biotechnology and Food Science 2019; 8(4), 1021-1025.
[24] Y Umidakhon, B Erkin, G Ulugbek, N Bahadir and A Karim. Correction of the mitochondrial NADH oxidase activity, peroxidation and phospholipid metabolism by haplogenin-7-glucoside in hypoxia and ischemia. Trends in Sciences 2022; 19(21), 6260.
[25] MK Pozilov, U Gayibov, MI Asrarov, NG Abdulladjanova, HS Ruziboev and TF Aripov. Physiological alterations of mitochondria under diabetes condition and its correction by polyphenol gossitan. Journal of Microbiology Biotechnology and Food Science 2022; 12(2), 2224.
[26] AG Vakhobjonovna, KE Jurayevich, AIZ Ogli, EN Azamovich, MR Rasuljonovich, and AM Islomovich. Tannins as modulators in the prevention of mitochondrial dysfunction. Trends in Sciences 2025; 22(8), 10436.
[27] Z Shakiryanova, R Khegay, U Gayibov, A Saparbekova, Z Konarbayeva, A Latif and O Smirnova. Isolation and study of a bioactive extract enriched with anthocyanin from red grape pomace (Cabernet Sauvignon). Agronomy Research 2023; 21(3), 1293-1303.
[28] U Gayibov, SN Gayibova, KP Ma’murjon, FS Tuxtaeva, UR Yusupova, GMK Djabbarova, ZA Mamatova, NA Ergashev and TF Aripov. Influence of quercetin and dihydroquercetin on some functional parameters of rat liver mitochondria. Journal of Microbiology, Biotechnology and Food Sciences 2021; 11(1), 2924.