Trends
Sci.
2026;
23(5):
11952
Histone Methyltransferase and Demethylase Gene in Pediatric Acute Lymphoblastic Leukemia: A Molecular Insights
Nur Aziz1, Yudha Nur Patria2, Amirah Ellyza Wahdi4,
Eddy Supriyadi2,3 and Dewajani Purnomosari1,*
1Department of Histology and Cell Biology, Faculty of Medicine, Public Health and Nursing,
Universitas Gadjah Mada, Yogyakarta 55281, Indonesia
2Department of Child Health, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada,
Yogyakarta 55281, Indonesia
3Dr. Sardjito General Hospital, Jalan Kesehatan, Yogyakarta 55281, Indonesia
4Department of Biostatistics, Epidemiology and Population Health, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada, Yogyakarta 55281, Indonesia
(*Corresponding author’s e-mail: [email protected])
Received: 26 September 2025, Revised: 29 October 2025, Accepted: 10 November 2025, Published: 1 January 2026
Abstract
Genetic factors driving the development of pediatric Acute Lymphoblastic Leukemia (ALL) remain incompletely understood. While epigenetic dysregulation through histone methyltransferases and demethylases has emerged as a critical oncogenic mechanism, their specific contributions to pediatric ALL require systematic investigation. In this study, we systematically investigated genetic variants in histone methyltransferases and demethylase found in top 50 mutated genes from TARGET ALL Phase 2 cohort. Single nucleotide variations were functionally annotated using PredictSNP and I-Mutant2.0, followed by survival analysis and pathway enrichment studies comparing mutated versus wild-type patients. KMT2D, NSD2, SETD2, EZH2 (methyltransferases), and KDM6A (demethylase), ranked among the most frequently mutated genes. Multiple frameshift and nonsense mutations were identified in these genes, likely resulting in truncated proteins and loss of function. Critically, missense variants in EZH2 (F145C, R679C, P132T, D652G, A576D, S280C, Y728H, C566S), KDM6A (G1242D), NSD2 (E1099K, D1125H), and SETD2 (G2170D, R1592Q) were computationally predicted as deleterious and destabilizing. Although survival analysis revealed no statistically significant differences (p = 0.37), mutation carriers exhibited a 44% increased mortality risk, suggesting clinical relevance despite limited sample size (n = 35). Gene set enrichment analysis of differentially expressed genes revealed significant activation of hematopoietic cell lineage pathways in mutated patients. Our findings underscore the importance of histone methyltransferase and demethylase gene variants in pediatric ALL, particularly novel mutations affecting protein function and stability that remain poorly characterized in the literature. This study identifies candidate mutations that warrant further functional and clinical investigation for their role in disease progression and therapeutic targeting.
Keywords: Epigenetic regulation, Gene mutation, Histone demethylase, Histone methyltransferase, Pediatric ALL
Introduction
Acute lymphoblastic leukemia (ALL) is the predominant cancer in children, accounting for approximately 75% - 80% of all pediatric acute leukemia cases [1,2]. This disease is characterized by the uncontrolled proliferation of immature lymphoid progenitor cells in the bone marrow and extramedullary sites, disrupting normal blood cell production and causing severe clinical symptoms [3,4]. While advancements in treatment have improved 5-year survival rates [5], pediatric ALL remains a biologically diverse disease with genetically and phenotypically heterogeneous leukemic subclones [6]. In particular, the genetic factors driving its development are still not fully understood. Identifying the genetic variants involved in pediatric ALL is critical for uncovering new therapeutic targets and improving outcomes for affected children.
Recent research has increasingly highlighted that genetic variations influence cancer risk and shape tumor mutational profiles [7], particularly through their impact on key cellular processes such as chromatin modification and epigenetic regulation [8]. Protein methyltransferases and demethylases are critical enzymes central to this epigenetic machinery, modulating the addition and removal of methyl groups on histone and non-histone proteins [9, 10]. These modifications are crucial for regulating gene expression, maintaining chromatin structure, and repairing DNA, all processes commonly disrupted in cancer. Several proteins in this class have been reported to be involved in ALL regulation, such as KMT2A [11,12], PRMT7 [13], EHMT2 [14], SETD2 [15], NSD2 [16,17]. Mutations in these genes often lead to loss of function, abnormal methylation patterns, or oncogenic transformation.
Despite growing evidence of their role in adult cancers, the impact of protein methyltransferase and demethylase gene variants in pediatric ALL remains largely unexplored. To date, no comprehensive studies have evaluated the functional and clinical implications of these genetic alterations of protein methyltransferases and demethylases in pediatric leukemia. This knowledge gap is significant, as these genes may play a key role in the epigenetic disruptions that drive pediatric ALL.
This study aims to systematically investigate the genetic variants in protein methyltransferase and demethylase genes in pediatric ALL. By examining their mutation spectrum, functional consequences, and potential effects on protein stability, we hope to shed light on the roles of protein methyltransferase and demethylase genes in pediatric ALL. Our findings could provide valuable insights into the epigenetic mechanisms underlying pediatric ALL and identify potential therapeutic targets or biomarkers for disease progression.
Materials and methods
Data collection and retrieval
The Simple Nucleotide Variation (SNV) data for the TARGET Acute Lymphoblastic Leukemia (ALL) Phase 2 project [18] was retrieved using the TCGAbiolinks R package, a bioinformatics tool that enables direct access to the NCI Genomic Data Commons (GDC) for querying, downloading, and performing integrative analyses [19, 20]. The data was filtered based on the workflow type “Aliquot Ensemble Somatic Variant Merging and Masking” to ensure high-quality somatic variant calls. Following retrieval, the Mutation Annotation Format (MAF) file was processed using the maftools package to identify the top 50 most frequently mutated genes. From this list, genes belonging to the protein methyltransferase and demethylase families were manually curated based on prior literature and functional relevance, including genes such as KMT2D, NSD2, SETD2, EZH2, and KDM6A. The resulting filtered data was visualized using an oncoplot to highlight mutation frequencies and patterns in the selected genes. All data retrieval, processing, and visualization steps were conducted in R, ensuring reproducibility and consistency throughout the analysis.
Functional prediction of missense variants
To evaluate the functional impact of missense mutations in key genes: KMT2D (O14686), NSD2 (O96028), SETD2 (Q9BYW2), EZH2 (Q15910), and KDM6A (O15550), we utilized the PredictSNP platform (https://loschmidt.chemi.muni.cz/predictsnp1/). The amino acid sequences for each gene were retrieved in FASTA format from the UniProt database using their respective accession IDs. The input data included the amino acid position and substitution for each missense mutation.
Protein stability prediction of missense variants
To predict the impact of missense mutations on protein stability, we utilized the I-Mutant2.0 tool [21], a widely used support vector machine (SVM)-based algorithm [22]. I-Mutant2.0 predicts changes in protein stability upon single amino acid substitutions by calculating the ΔΔG values, which represent the change in Gibbs free energy. Negative ΔΔG values indicate a decrease in protein stability, whereas positive values suggest an increase in stability. Each missense variant was individually input into I-Mutant2.0 along with the wild-type protein sequence. Predictions were performed under default conditions, assuming a temperature of 25 °C and pH of 7.0, to simulate physiological conditions.
Survival analysis of ALL patients with methyltransferase/demethylase gene mutations
To evaluate the clinical significance of methyltransferase/demethylase gene alterations pediatric ALL, we conducted a retrospective survival analysis using genomic and clinical datasets from the TARGET-ALL Phase II Discovery cohort, obtained through the Genomic Data Commons via the TCGAbiolinks R package. We examined somatic mutations of SETD2, EZH2, KMT2D, KDM6A, and NSD2. Mutation data were analyzed using the maftools R package, with inclusion criteria restricted to functionally impactful variants including missense mutations, nonsense mutations, frameshift deletions and insertions, splice site alterations, and in-frame insertions/deletions. Study participants were stratified into binary categories based on their mutational profile: “Non-mutated” (absence of functionally relevant mutations across all 5 target genes) and “Mutated” (detection of one or more functionally relevant mutations in any target gene). Patient clinical characteristics were obtained from the TARGET-ALL Clinical Data Supplement and integrated with genomic data using unique patient identifiers.
Survival analysis was performed using Kaplan-Meier methods with overall survival as the primary endpoint, defined as time from diagnosis to death from any cause, with surviving patients censored at last follow-up. Survival distributions between mutated and non-mutated groups were compared using the log-rank test, and hazard ratios with 95% confidence intervals were calculated using univariate Cox proportional hazards regression. Only patients with complete survival data (non-missing survival time, event status, and survival time > 0 days) and definitive mutation status were included in the analysis. All statistical analyses were performed using R with survival and survminer packages [23], with statistical significance set at α = 0.05.
Exploratory gene expression analysis of ALL patients with methyltransferase/demethylase gene mutations
RNA-seq gene expression data for the TARGET-ALL Phase II cohort were obtained from the Genomic Data Commons using TCGAbiolinks R package, specifically querying transcriptome profiling data generated through the STAR-Counts workflow from primary blood-derived cancer samples. Expression count matrices were merged with clinical data and mutation status information, resulting in a filtered dataset of samples with concurrent genomic, transcriptomic, and clinical data availability. Samples were stratified into mutated and non-mutated groups based on the presence of functionally impactful variants in any of the 5 target methyltransferase/demethylase genes (SETD2, EZH2, KMT2D, KDM6A, NSD2) as previously defined. Only samples with complete mutation status classification and RNA-seq data were retained for differential expression analysis.
Differential gene expression analysis was performed using the limma-voom pipeline in R. Raw count data were processed using DGEList from the edgeR package [24], followed by normalization factor calculation and voom transformation using the limma pipeline, which effectively accounts for mean-variance relationships and has been proven superior for RNA-seq data analysis [25,26]. A linear model was fitted using mutation status as the primary factor, with empirical Bayes moderation applied to improve statistical inference. Ensembl gene identifiers were converted to HGNC gene symbols using the org.Hs.eg.db annotation package, with duplicate symbols resolved by selecting the transcript with the lowest adjusted p-value. Differentially expressed genes were classified as upregulated (log2 fold change > 1, adjusted p-value < 0.05), downregulated (log2 fold change < −1, adjusted p-value < 0.05), or non-significant based on established thresholds, with false discovery rate correction applied using the Benjamini-Hochberg method.
Expression patterns were visualized using hierarchical clustering heatmaps generated with ComplexHeatmap package [27], displaying Z-score normalized expression values for the top differentially expressed genes with mutation status annotation. Gene Set Enrichment Analysis (GSEA) was conducted using the clusterProfiler package [28] to identify significantly enriched KEGG pathways, employing a ranked gene list based on log2 fold change values without applying arbitrary cutoffs. All genes were ranked from highest to lowest fold change, with ENTREZ gene identifiers used for pathway mapping and results converted to gene symbols for interpretation. GSEA parameters included minimum gene set size of 15, maximum of 500, and FDR-adjusted p-value cutoff of 0.05.
Pathway-specific gene analysis focused on the significantly enriched pathways, extracting the 8 most influential genes per pathway based on core enrichment and absolute log2 fold change rankings. Gene-gene interaction networks were constructed to visualize shared pathway memberships, with nodes representing genes and edges indicating co-occurrence across multiple pathways. Network analysis was performed using igraph [28] packages, with node sizes reflecting pathway involvement frequency and edge weights representing the number of shared pathways between connected genes.
Results and discussion
Genetic alterations in protein methyltransferases and demethylases genes in pediatric ALL
From the analysis of the 50 most frequently mutated genes in the TARGET ALL Phase 2 cohort (Figure 1(A) and Table S1), 5 genes of interest were identified: KMT2D, NSD2, SETD2, and EZH2, which encode protein methyltransferases, and KDM6A, which encodes a protein demethylase. These genes are critical for epigenetic regulation, playing essential roles in chromatin remodeling and gene expression control. Mutations in these genes were observed in a substantial proportion of the samples, with their frequencies shown in Figure 1(B). Despite their potential importance, the role of these genetic alterations remains largely unexplored. This finding underscores the need to investigate the genetic variants observed in pediatric ALL patients to uncover potential driver mutations or deleterious variants that may contribute to disease initiation or progression.
Among the 93 samples harboring mutations in any of the methyltransferase/demethylase genes, KMT2D demonstrated the highest alteration frequency, being mutated in 32 samples (34%), encompassing a diverse spectrum of variant types including missense, nonsense, and frameshift mutations. NSD2 ranked second with mutations in 23 samples (25%), characterized predominantly by missense variants. SETD2 exhibited alterations in 19 samples (20%), including splice site, nonsense, missense, and frameshift mutations. EZH2 mutations occurred in 14 samples (15%) and were primarily composed of missense alterations, while KDM6A showed the lowest frequency with mutations in 12 samples (13%), displaying a heterogeneous pattern of missense, frameshift, and nonsense variants. Although mutations were identified in 93 samples, they corresponded to only 89 unique patients, as some individuals had multiple samples analyzed. Taken together, this mutational landscape underscores KMT2D as the predominant target of genetic disruption in pediatric ALL and highlights the differential vulnerability of methyltransferase and demethylase genes to distinct types of genetic alterations.
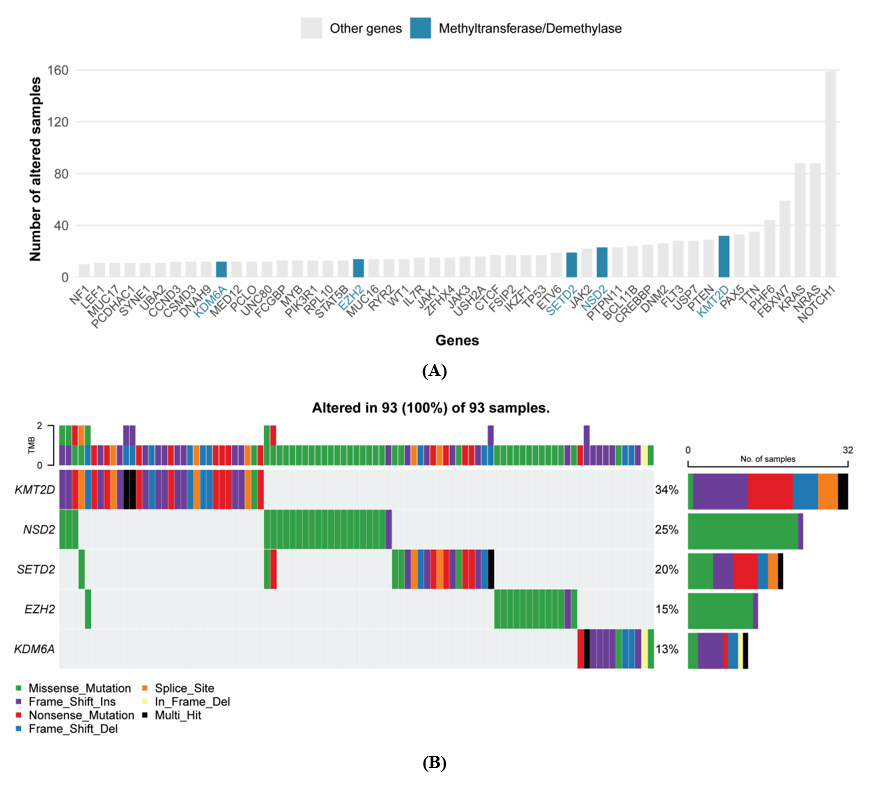
Figure 1 Genetic alterations in protein methyltransferases and demethylases genes in TARGET ALL phase 2 cohort. (A) The bar plot shows the top 50 mutated genes in TARGET ALL Phase 2, with protein methyltransferase/demethylase genes highlighted in blue, while other genes are in gray. The y-axis represents the number of samples altered. (B) The oncoplot shows the mutation distribution of 5 protein methyltransferase/demethylase genes (KMT2D, NSD2, SETD2, EZH2, KDM6A), with KMT2D being the most frequently mutated (34%) across 93 samples.
Truncating mutations in methyltransferases and demethylases genes
Frameshift and mutations identified in methyltransferases and demethylases genes were provided in Table 1. Frameshift mutations were identified in key protein methyltransferase and demethylase genes. KMT2D exhibited the highest frequency of alterations, with frameshift insertions/deletions and nonsense mutation such as p.K287Dfs*2, p.T382Hfs*2, p.Q170Afs*49, and p.G2219_A2220insVR*, distributed across its coding regions. KDM6A, positioned on the X chromosome, exhibited clustered frameshift insertions and deletions within a narrow genomic region, including p.R1111Gfs40, p.V1112Dfs38, and p.V1113Gfs*5, indicative of a mutational hotspot prone to genetic instability. A single nonsense mutation (p.W355*) was identified in KDM6A, likely leading to loss of its demethylase function. Notably, frameshift mutations at residue R1111 were detected in 2 patients, while alterations at V1113 were observed in 3 patients. The concentration of frameshift variants within consecutive amino acid positions (1111 -1 113) implies that this domain may harbor critical functional motifs essential for KDM6A’s demethylase activity, making it a vulnerable target for loss-of-function alterations in pediatric ALL pathogenesis. SETD2 demonstrated additional truncating alterations, such as frameshift mutation p.A1851Vfs*15 and nonsense mutation p.E1115*. In contrast, NSD2 exhibited a more restricted truncating mutation profile, with only a single frameshift insertion p.F718Lfs*6 identified. Similarly, EZH2 showed minimal truncating disruption with a solitary frameshift insertion p.K735Efs*23.
These findings highlight a substantial truncating mutations affecting 44 pediatric ALL patients, with KMT2D emerging as the predominant target of genetic disruption, harboring frameshift and nonsense mutations in 23 patients. Remarkably, 26 distinct truncating mutations were classified as novel or absent from dbSNP databases, indicating previously uncharacterized genetic alterations specific to pediatric ALL and highlighting the critical need for expanded mutational databases and functional validation studies. Collectively, these results underscore the fundamental role of methyltransferase and demethylase gene disruption in pediatric ALL, with truncating mutations representing a major mechanism of epigenetic dysregulation that warrants further investigation as potential therapeutic targets and prognostic biomarkers.
Table 1 Frameshift and nonsense mutation in methyltransferase and demethylase genes.
No. |
Hugo Symbol |
Chromosome |
Variant Classification |
HGVSp Short |
dbSNP RS |
Sample ID |
1 |
EZH2 |
chr7 |
Frame Shift Ins |
p.K735Efs*23 |
novel |
TARGET-10-PATWNL |
2 |
KDM6A |
chrX |
Frame Shift Del |
p.V1113Gfs*5 |
novel |
TARGET-10-PASXIL |
3 |
KDM6A |
chrX |
Frame Shift Ins |
p.V1113Ffs*10 |
NA |
TARGET-10-PASYSJ |
4 |
KDM6A |
chrX |
Frame Shift Del |
p.V1112Dfs*38 |
novel |
TARGET-10-PASMNV |
5 |
KDM6A |
chrX |
Frame Shift Ins |
p.V1113Gfs*40 |
novel |
TARGET-10-PASJJR |
6 |
KDM6A |
chrX |
Frame Shift Ins |
p.G58Rfs*6 |
NA |
TARGET-10-PARMWH |
7 |
KDM6A |
chrX |
Frame Shift Ins |
p.M1291Ifs*3 |
novel |
TARGET-10-PASGFH |
8 |
KDM6A |
chrX |
Frame Shift Ins |
p.R1111Afs*40 |
novel |
TARGET-10-PARDWE |
9 |
KDM6A |
chrX |
Frame Shift Ins |
p.R1111Gfs*40 |
NA |
TARGET-10-PARPYJ |
10 |
KDM6A |
chrX |
Nonsense Mutation |
p.W355* |
NA |
TARGET-10-PARDLR |
11 |
KMT2D |
chr12 |
Frame Shift Ins |
p.K287Dfs*2 |
NA |
TARGET-10-PAPEFH |
12 |
KMT2D |
chr12 |
Frame Shift Ins |
p.L1461Tfs*30 |
novel |
TARGET-10-PARGFL |
13 |
KMT2D |
chr12 |
Frame Shift Del |
p.S1476Lfs*14 |
novel |
TARGET-10-PARGJY |
14 |
KMT2D |
chr12 |
Frame Shift Del |
p.A5042Pfs*9 |
novel |
TARGET-10-PARBNY |
15 |
KMT2D |
chr12 |
Frame Shift Ins |
p.G1758Pfs*29 |
novel |
TARGET-10-PARTAK |
16 |
KMT2D |
chr12 |
Frame Shift Del |
p.R4546Gfs*28 |
novel |
TARGET-10-PARIIA |
17 |
KMT2D |
chr12 |
Frame Shift Ins |
p.L752Rfs*6 |
novel |
TARGET-10-PARBXX |
18 |
KMT2D |
chr12 |
Frame Shift Del |
p.Y5317Ffs*27 |
novel |
TARGET-10-PARJLF |
19 |
KMT2D |
chr12 |
Frame Shift Ins |
p.R3539Dfs*121 |
novel |
TARGET-10-PAPLDL |
20 |
KMT2D |
chr12 |
Frame Shift Ins |
p.S5201Efs*43 |
novel |
TARGET-10-PARCDS |
21 |
KMT2D |
chr12 |
Frame Shift Ins |
p.E3504Gfs*4 |
novel |
TARGET-10-PANTWC |
22 |
KMT2D |
chr12 |
Frame Shift Ins |
p.L2689Pfs*4 |
novel |
TARGET-10-PAREYW |
23 |
KMT2D |
chr12 |
Frame Shift Ins |
p.S3713Lfs*38 |
novel |
TARGET-10-PARBLL |
24 |
KMT2D |
chr12 |
Frame Shift Ins |
p.L3042Pfs*8 |
novel |
TARGET-10-PASJMK |
25 |
KMT2D |
chr12 |
Frame Shift Ins |
p.T382Hfs*2 |
novel |
TARGET-10-PAPNNX |
26 |
KMT2D |
chr12 |
Nonsense Mutation |
p.Q3767* |
novel |
TARGET-10-PARBLS |
27 |
KMT2D |
chr12 |
Nonsense Mutation |
p.E3056* |
NA |
TARGET-10-PARMKK |
28 |
KMT2D |
chr12 |
Nonsense Mutation |
p.R5454* |
rs267607239 |
TARGET-10-PAPCUI |
29 |
KMT2D |
chr12 |
Nonsense Mutation |
p.Q5261* |
NA |
TARGET-10-PANTTB |
30 |
KMT2D |
chr12 |
Nonsense Mutation |
p.R2635* |
rs794727549 |
TARGET-10-PARCLU |
31 |
KMT2D |
chr12 |
Nonsense Mutation |
p.W1491* |
NA |
TARGET-10-PAPSPN |
32 |
KMT2D |
chr12 |
Nonsense Mutation |
p.R5097* |
NA |
TARGET-10-PATZFF |
33 |
KMT2D |
chr12 |
Nonsense Mutation |
p.G2219_A2220insVR* |
NA |
TARGET-10-PARJZZ |
34 |
NSD2 |
chr4 |
Frame Shift Ins |
p.F718Lfs*6 |
novel |
TARGET-10-PASXSI |
35 |
SETD2 |
chr3 |
Frame Shift Ins |
p.A1851Vfs*15 |
novel |
TARGET-10-PARGHW |
36 |
SETD2 |
chr3 |
Frame Shift Ins |
p.T2388Nfs*41 |
rs1323986990 |
TARGET-10-PAPAZD |
37 |
SETD2 |
chr3 |
Frame Shift Del |
p.Q2342Kfs*11 |
novel |
TARGET-10-PAPAMS |
38 |
SETD2 |
chr3 |
Frame Shift Ins |
p.T1269Lfs*64 |
novel |
TARGET-10-PAPAGV |
39 |
SETD2 |
chr3 |
Frame Shift Ins |
p.E1922Vfs*24 |
NA |
TARGET-10-PATXKW |
40 |
SETD2 |
chr3 |
Frame Shift Del |
p.K1426Lfs*5 |
novel |
TARGET-10-PATGYH |
41 |
SETD2 |
chr3 |
Frame Shift Ins |
p.L173Sfs*64 |
NA |
TARGET-10-PATDRC |
42 |
SETD2 |
chr3 |
Nonsense Mutation |
p.W1306* |
NA |
TARGET-10-PARTGB |
43 |
SETD2 |
chr3 |
Nonsense Mutation |
p.R70* |
rs775039657 |
TARGET-10-PAPFNV |
44 |
SETD2 |
chr3 |
Nonsense Mutation |
p.E1115* |
NA |
TARGET-10-PARKZX |
Deleterious missense mutation prediction
Missense mutations of KMT2D, SETD2, NSD2, EZH2, and KDM6A identified in TARGET ALL Phase 2 cohort were provided in Table S2. Critically, recurrent mutation hotspots emerged, including NSD2 E1099K in 11 patients representing the most prevalent single amino acid substitution, alongside additional NSD2 hotspots at D1125 (3 patients) and T1150A (2 patients). Meanwhile, EZH2 R679 missense mutation were also found in 2 patients. These recurrent missense mutations at specific amino acid residues suggest strong selective pressure for disrupting particular functional domains, emphasizing their potential as driver mutations in pediatric ALL pathogenesis and highlighting NSD2 E1099K as a predominant mutational event warranting immediate functional characterization and therapeutic targeting.
Missense mutations in KMT2D, SETD2, NSD2, EZH2, and KDM6A were evaluated using multiple in silico prediction tools such as PredictSNP, MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, SNAP, and PANTHER to assess their potential deleterious impact. Each prediction tool used in this study employs distinct algorithms to assess the functional impact of missense mutations, providing complementary insights into variant pathogenicity. PredictSNP, the most robust and reliable tool, integrates results from 6 individual predictors (MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, and SNAP) into a consensus classifier to determine whether an amino acid substitution is neutral or deleterious, improving accuracy by reducing biases and discrepancies inherent to single methods [29,30]. Multivariate Analysis of Protein Polymorphism (MAPP) analyzes evolutionary conservation and physicochemical properties like charge and polarity to identify disruptive substitutions [31]. Predictor of human Deleterious Single Nucleotide Polymorphisms (PhD-SNP) uses a machine learning-based model trained on sequence features to classify variants as neutral or disease-causing [32]. Polymorphism Phenotyping (Polyphen-1 and PolyPhen-2) combine sequence conservation and structural information, with PolyPhen-2 providing refined probabilistic scores [33]. Sorting Intolerant From Tolerant (SIFT) relies on sequence homology to identify mutations at highly conserved sites, classifying scores below 0.05 as damaging [34,35]. Screening for Non-Acceptable Polymorphisms (SNAP) employs a neural network to integrate evolutionary data and functional annotations for binary classification [36], while PANTHER uses evolutionary relationships and protein family annotations to determine the likelihood of functional disruption [37]. Together, these tools offer a multi-dimensional analysis of missense mutations, with PredictSNP emerging as the most comprehensive predictor due to its ability to consolidate individual outputs into high-confidence consensus scores.
Based on the in silico predictions as in Figure 2, several missense mutations were consistently classified as deleterious across multiple tools, making them strong candidates for further functional studies. In KMT2D, the variant C1400Y was predicted as highly deleterious in all available prediction tools. For SETD2 mutations, R1592Q, R1686W, H2514R, and G2170D were identified as potentially disruptive, with scores consistently exceeding 0.7 in PredictSNP in the case of R1592Q and R1686W. In NSD2, variants E1099K, D1125H, and D1009H stood out, particularly in PredictSNP, PhD-SNP, PolyPhen-2 and SIFT, suggesting significant functional impact.
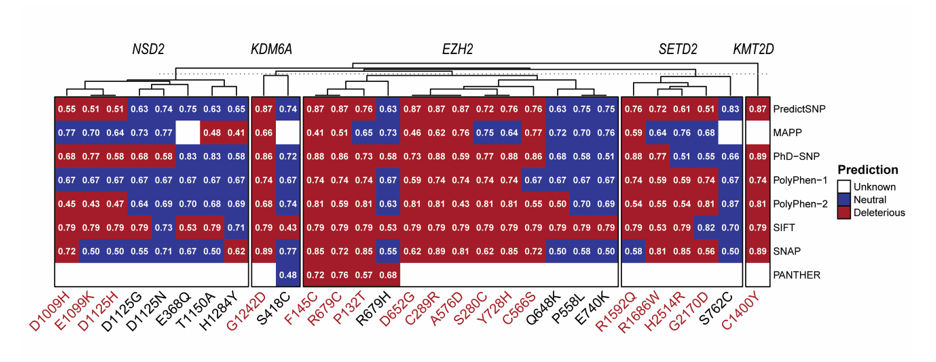
Figure 2 Computational prediction for the functional disruption of missense SNPs in protein methyltransferases and demethylase genes. Red cells indicate predicted as deleterious, blue cells represent neutral predictions, and white cells denote missing or non-applicable data (unknown). Higher scores reflect stronger confidence in deleterious/neutral classification.
In EZH2, the mutations F145C, R679C, P132T, D652G, C289R, A576D, S280C, Y728H, and C566S were strongly predicted to be deleterious, with high consensus scores in almost all prediction tools utilized, indicating their likely role in disrupting EZH2’s activity. Finally, in KDM6A, the variant G1242D consistently scored highly across PredictSNP, MAPP, PhD-SNP, Polyphen-1 and -2, SIFT, and SNAP highlighting its potential to impair demethylase function. These deleterious mutations across KMT2D, SETD2, NSD2, EZH2, and KDM6A represent promising candidates for further experimental validation to elucidate their impact on epigenetic regulation and their role in the pathogenesis of pediatric ALL.
Impact prediction of missense mutations on protein stability
To evaluate the effect of missense mutations on protein stability, we employed the I-Mutant2.0 prediction tool, a support vector machine (SVM)-based algorithm that predicts the impact of amino acid substitutions on protein stability [21]. I-Mutant2.0 uses sequence and structural information to provide a ΔΔG score, where negative values indicate a decrease in protein stability, and positive values signify increased stability [21]. This tool is widely used for assessing the consequences of mutations, as protein stability is critical for maintaining proper function and interactions.
The previously identified and predicted deleterious missense SNPs were further analyzed to assess their impact on protein stability using I-Mutant2.0. The results presented in Figure 3 show that several mutations are predicted to significantly alter protein stability across SETD2, NSD2, KMT2D, KDM6A, and EZH2. Gene-specific analysis demonstrated considerable variation in the distribution of predicted stability effects, with most of genes except of KMT2D showing predominantly destabilizing mutations (Figure 3(A)). Individual mutation assessment revealed a spectrum of stability impacts, ranging from highly destabilizing mutation EZH2 D652G (ΔΔG = −2.45 kcal/mol) to mildly stabilizing variant KMT2D (ΔΔG = 0.50 kcal/mol), with a median ΔΔG value of −0.97 kcal/mol indicating an overall destabilizing trend (Figure 3(B)). Gene-wise profiling revealed important differences in the mutational impact patterns among methyltransferases/demethylase (Figure 3(C)).
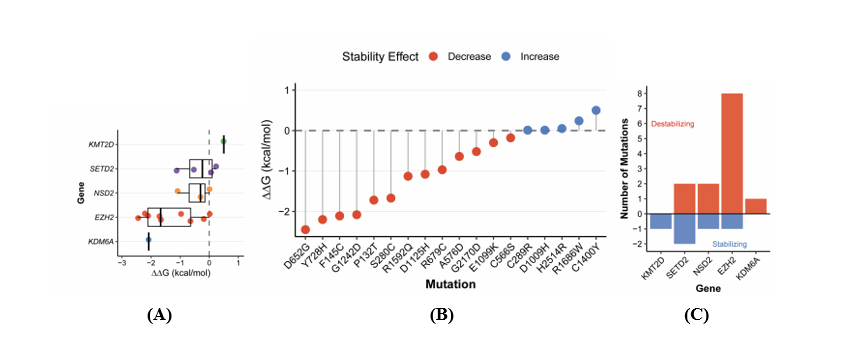
Figure 3 Computational prediction of protein stability changes induced by missense mutations. (A) Gene-specific distribution of predicted stability changes (ΔΔG values) showing median values and interquartile ranges with individual mutation effects overlaid as jittered points. Genes are ordered by median ΔΔG values. (B) Individual mutation effects ranked by predicted stability impact, displayed as a lollipop plot with mutations ordered from most destabilizing to most stabilizing. (C) Gene-wise summary showing the absolute number of destabilizing (positive bars, red) and stabilizing mutations (negative bars, blue) for each gene. The dashed horizontal line at ΔΔG = 0 indicates the stability threshold. Red coloring indicates destabilizing mutations (ΔΔG < 0 kcal/mol), while blue indicates stabilizing mutations (ΔΔG > 0 kcal/mol).
EZH2 demonstrated the most severe destabilizing profile with 8 out of 9 mutations predicted to compromise protein stability. EZH2, a core component of the PRC2 chromatin-modifying complex, catalyzes H3K27 methylation to regulate chromatin compaction and transcriptional repression [38]. Deleterious variants (F145C, R679C, P132T, D652G, A576D, S280C, Y728H and C566S) were further predicted to compromise protein structural stability, suggesting a dual mechanism of EZH2 inactivation through both catalytic disruption and protein destabilization. Among the most notable findings, the D652G and Y728H mutations in EZH2 show the largest decrease in protein stability, with I-Mutant ΔΔG scores of approximately −2.45 and −2.2 kcal/mol, respectively. These destabilizing mutations may impair EZH2’s methyltransferase activity, which is critical for histone H3 lysine 27 trimethylation (H3K27me3) and gene repression.
In KDM6A, the single mutation G1242D also led to a significant decrease in stability (ΔΔG = −2.08 kcal/mol), potentially affecting its demethylase function in removing H3K27 methylation. This loss of stability could disrupt the delicate balance of methylation-demethylation dynamics, leading to epigenetic dysregulation. KDM6A is expressed in hematopoietic stem and progenitor cells (HSPCs) [39], where it acts as a critical regulator of HSPC homeostasis [39,40], suggesting that its disruption may contribute to aberrant hematopoietic differentiation. Of particular interest, we identified a frameshift mutational hotspot within KDM6A spanning consecutive amino acid positions 1,111 – 1,113, indicating a potential vulnerability region that may be preferentially targeted during leukemic transformation.
Similarly, the R1592Q mutation in SETD2, the sole enzyme responsible for transcription-coupled histone H3 lysine 36 trimethylation (H3K36me3), markedly decreases protein stability (−1.13 kcal/mol), likely impairing its critical role in transcriptional regulation and DNA repair [41]. In NSD2, the D1125H mutation led to a significant decrease in stability (ΔΔG = −1.08), which could impair its histone methyltransferase activity, particularly toward its known substrate, histone H3 lysine 36 (H3K36me2), as well as its role in chromatin remodeling [42,43]. Interestingly, KMT2D harbored the C1400Y mutation, resulting in a slight increase in protein stability (ΔΔG = 0.5 kcal/mol). Although stabilizing mutations are less common, they can still disrupt protein function by affecting flexibility or interaction dynamics. As a histone H3 lysine 4 (H3K4) methyltransferase, alterations in KMT2D can trigger aberrant epigenomic reprogramming and consequent reconfiguration of molecular pathways [44].
Overall, these results highlight key destabilizing mutations in EZH2 (F145C, R679C, P132T, D652G, A576D, S280C, Y728H, C566S), KDM6A (G1242D), NSD2 (E1099K, D1125H), and SETD2 (G2170D, R1592Q). These mutations are likely to disrupt protein function, leading to downstream consequences in epigenetic regulation and transcriptional control. Conversely, the stabilizing mutation in KMT2D (C1400Y) warrants further investigation to understand its biological implications. Experimental validation of these findings will provide critical insights into the functional impact of these variants in the context of disease.
Survival analysis of all patients with mutations in methyltransferase/demethylase genes
Our analysis identified 93 samples harboring mutations in one or more of the target epigenetic regulatory genes (KMT2D, NSD2, SETD2, EZH2, and KDM6A) based on oncoplot visualization (Figure 1(B)). Among these, 89 patients carried functionally impactful variants. After applying stringent data quality filters requiring complete survival information and definitive mutation status, the final analytical cohort comprised of 35 patients: 24 non-mutated (68.6%) and 11 mutated (31.4%) cases.
Univariate Cox proportional hazards analysis revealed that patients with methyltransferase/demethylase gene mutations had a hazard ratio of 1.448 (95% CI: 0.641 - 3.274, p = 0.3733) compared to non-mutated patients. Although this association did not reach statistical significance, the hazard ratio indicates a 44.8% increased risk of death among mutated patients, suggesting a potential adverse prognostic trend. This clinically meaningful effect size warrants further investigation in larger cohorts with enhanced statistical power to definitively establish the prognostic significance of these epigenetic alterations. Median overall survival analysis supported this trend, with non-mutated patients demonstrating longer survival (1,074 days; 95% CI: 645 - 1,752 days) compared to mutated patients (846 days; 95% CI: 510-not reached) (Figure 4). This represents an absolute difference of 228 days (approximately 7.5 months) in median survival, though the confidence intervals overlapped considerably. The upper confidence limit for the mutated group could not be estimated due to insufficient events, reflecting the limited sample size and highlighting the need for validation in expanded patient cohorts to confirm these preliminary survival differences.
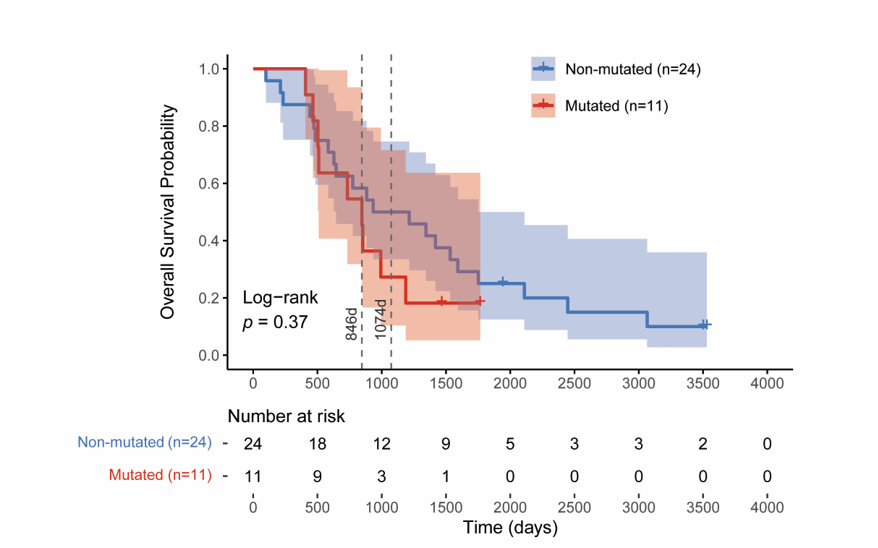
Figure 4 Kaplan-Meier survival analysis stratified by mutation status. Overall survival curves for patients with mutations in any of 5 chromatin-modifying genes (SETD2, NSD2, KMT2D, KDM6A, EZH2) versus wild-type patients. Median survival: 846 days (mutated) vs 1,047 days (wild-type/non-mutated). HR = 1.448 (95% CI: 0.641 - 3.274, p = 0.3733, log-rank test).
Exploratory gene expression analysis of ALL patients with methyltransferase/demethylase gene mutations
Integration of RNA-seq expression data with clinical and mutation information yielded a highly limited analytical cohort of only 5 patients, comprising 3 mutated and 2 non-mutated cases. The mutated patients harbored functionally impactful variants in 3 different genes: one patient (TARGET-10-PAPLDL) carried a frameshift insertion in KMT2D (p.R3539Dfs*121, chr12:49034193-49034194), another patient (TARGET-10-PARIAD) had a missense mutation in NSD2 (p.E1099K, chr4:1961074), and the third patient (TARGET-10-PARDWE) presented with 2 frameshift insertions in KDM6A (p.R1111Afs*40, chrX:45083504-45083505 and p.R1111Efs*40, chrX:45083506). The remaining 2 patients (TARGET-10-PASFXA and TARGET-10-PAKSWW) showed no mutations in the target genes.
Differential gene expression analysis identified minimal transcriptomic differences between mutated and non-mutated groups, with only 80 genes (0.22%) showing significant differential expression at the applied thresholds (|log2FC| > 1, adjusted p-value < 0.05). Among these, 50 genes were upregulated and 30 were downregulated in the mutated group, while 36,135 genes showed no significant changes (Figure 5A). The top 10 upregulated genes included BHLHE23, FLJ16779, NKAIN4, LINC00689, BEST3, IRX2, IGHA1, LDB3, IGKC, and PRKCG, representing diverse functional categories including transcriptional regulation (BHLHE23), immunoglobulin components (IGHA1, IGKC), and protein kinase signaling (PRKCG) (Figure 5(B)). Conversely, the most downregulated genes comprised F13A1, CACNA2D4, ANKRD30B, GSTM1, LRRC15, CFAP221, LOC124904634, TFAP2C, PSG4, and CDKN2A, encompassing roles in blood coagulation (F13A1), calcium signaling (CACNA2D4), detoxification (GSTM1), and cell cycle regulation (CDKN2A) (Figure 5(B)).
Gene Set Enrichment Analysis (GSEA) revealed several significantly enriched KEGG pathways. Top 5 positively enriched pathways included hematopoietic cell lineage (NES = 2.035, p.adjust = 3.63×10⁻⁴), Rap1 signaling pathway (NES = 1.836, p.adjust = 3.63×10⁻⁴), graft-versus-host disease (NES = 2.193, p.adjust = 8.88×10⁻⁴), sphingolipid signaling pathway (NES = 1.857, p.adjust = 1.38×10⁻³), and hippo signaling pathway (NES = 1.798, p.adjust = 1.38×10⁻³) (Figure 5(C)). The enrichment of hematopoietic cell lineage and graft-versus-host disease pathways underscores their relevance to ALL pathogenesis, suggesting that mutations in methyltransferase and demethylase genes, particularly KMT2D, NSD2, and KDM6A, may contribute to the dysregulation of hematopoietic differentiation and immune recognition processes.
Among the enriched KEGG pathways, the eight most influential genes demonstrated distinct expression patterns with significant biological implications (Figure 5(D)). PRKCG, encoding protein kinase C gamma, exhibited the highest upregulation with a 5.84-fold increase in the mutated group compared to non-mutated group, participating in both Rap1 and sphingolipid signaling pathways. Additionally, CR1L, IL1A, and HLA-DQA1 emerged as key drivers of the hematopoietic cell lineage pathway, displaying fold changes ranging from 3.32 to 3.5. Notably, HLA-DQA1 and IL1A established critical connections between hematopoietic cell lineage and graft-versus-host disease pathways (Figure 5(E)), although KIR2DL3 and KLRD1 represented the most influential genes within the latter.
Gene-gene interaction network analysis across the 5 enriched pathways revealed functionally significant connectivity patterns (Figure 5(E)). Two biologically meaningful connections emerged: The Rap1 and sphingolipid signaling pathways shared 3 genes (PLCB3, PRKCG, and GNAI1), all of which participate in intracellular signaling cascades, while PARD6G serves as a molecular link between the Rap1 and Hippo signaling pathways. Furthermore, hematopoietic cell lineage and graft-versus-host disease pathways converged through 2 shared genes (HLA-DQA1 and IL1A), both essential for immune recognition and inflammatory responses. HLA-DQA1 encodes an MHC class II molecule critical for antigen presentation, whereas IL1A functions as a pivotal pro-inflammatory cytokine, suggesting that epigenetic mutations may dysregulate immune surveillance mechanisms in pediatric ALL. These pathway interconnections, while requiring validation in expanded cohorts, provide mechanistic insights into how chromatin-modifying gene mutations potentially influence leukemogenesis through disrupted hematopoietic differentiation and compromised immune evasion pathways.
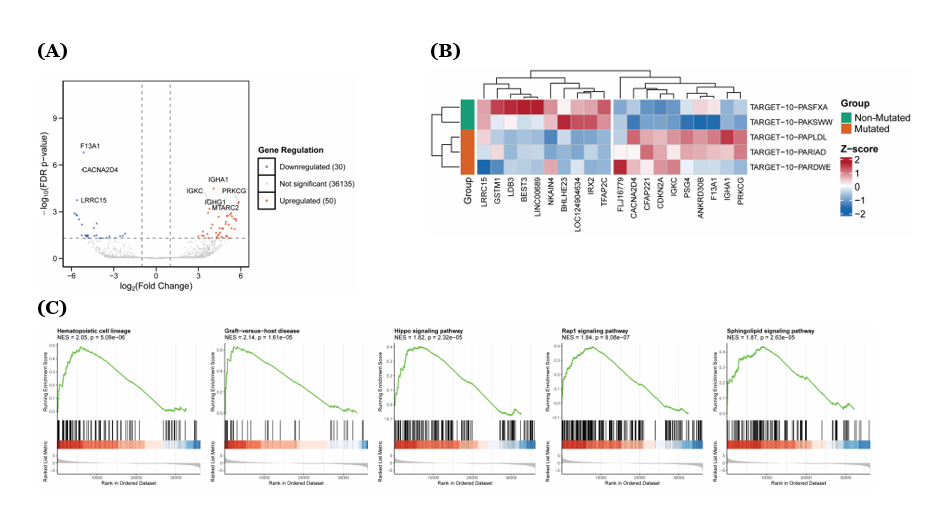
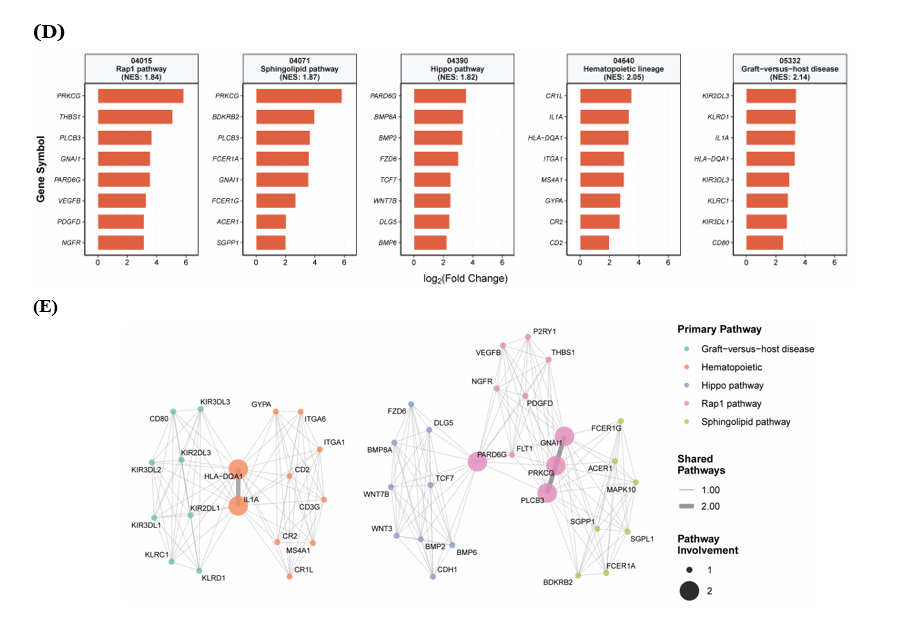
Figure 5 Exploratory analysis of differentially expressed genes between patients with and without methyltransferases/demethylase mutations. (A) Volcano plot showing log₂ fold change versus -log10 (FDR-adjusted p-value) for all analyzed genes. Red dots indicate significantly upregulated genes (log₂FC > 1, FDR < 0.05), blue dots represent significantly downregulated genes (log₂FC < −1, FDR < 0.05), and gray dots show non-significant genes. Dashed lines indicate significance thresholds. (B) Heatmap displaying Z-score normalized expression of the top 10 most significantly upregulated and downregulated genes. Samples are clustered by mutation status with color-coded annotations (green: Wild-type/non-mutated, orange: mutated) (C) Gene Set Enrichment Analysis (GSEA) results for KEGG pathways showing normalized enrichment scores (NES) and significance levels (p-value) for enriched pathways. (D) Bar plot of log₂ fold changes for the 8 most influential core enrichment genes within each significantly enriched pathway, ranked by absolute fold change magnitude. (E) Gene-gene interaction network displaying shared genes across enriched pathways. Node size represents the number of pathways each gene participates in, node color indicates the primary pathway assignment, and edge thickness represents the number of shared pathways between connected genes.
Conclusions
This comprehensive analysis reveals that methyltransferases and demethylase genes (EZH2, KDM6A, NSD2, SETD2 and KMT2D) are frequently mutated in pediatric ALL. KMT2D exhibited the highest mutation frequency predominantly through frameshift/nonsense alterations, while EZH2, KDM6A, NSD2, and SETD2 harbored multiple deleterious and destabilizing missense variants including EZH2 (F145C, R679C, P132T, D652G, A576D, S280C, Y728H, C566S), KDM6A (G1242D), NSD2 (E1099K, D1125H), and SETD2 (G2170D, R1592Q). While these mutations did not significantly impact overall survival in the current cohort, the observed trend toward poorer outcomes and associated alterations in immune-related gene expression suggest potential clinical relevance that warrants validation in larger studies. These findings underscore the potential role of these mutations in disrupting epigenetic regulation and contributing to leukemogenesis, warranting further functional validation to explore their significance in disease initiation and progression.
Acknowledgements
Thank you to Adrian Coen for proofreading the article, Bilqis Zahra Nabila for writing assistance and Penta Akhirul Awal for her administrative support.
Declaration of generative AI in scientific writing
The authors acknowledge the use of generative AI tools (ChatGPT) for language refinement and grammatical editing during the preparation of this manuscript. No content generation or data interpretation was performed by AI. The authors retain full responsibility for the integrity, content, and conclusions presented in this work.
CRediT author statement
Nur Aziz: Conceptualization; Methodology; Software; Investigation; Writing - Review & Editing. Yudha Nur Patria: Data curation; Validation; Formal analysis. Amirah Ellyza Wahdi: Data curation; Validation; Formal analysis. Eddy Supriyadi: Formal analysis; Supervision. Dewajani Purnomosari: Formal analysis; Supervision; Writing - Reviewing and Editing.
References
[1] H Inaba, D Teachey, C Annesley, S Batra, J Beck, S Colace, S Cooper, M Dallas, SD Oliveira, K Kelly, C Kitko, M Kohorst, M Kutny, N Lacayo, C Lee-Miller, K Ludwig, L Madden, K Maloney, D Mangum, …, K Stehman. Pediatric acute lymphoblastic leukemia, version 2.2025, NCCN clinical practice guidelines in oncology. Journal of the National Comprehensive Cancer Network 2025; 23(2), 41-62.
[2] A Mohammadian-Hafshejani, IM Farber and S Kheiri. Global incidence and mortality of childhood leukemia and its relationship with the Human Development Index. PLoS One 2024; 19(7), e0304354.
[3] MS Lajevardi, M Ashrafpour, SMH Mubarak, B Rafieyan, A Kiani, E Noori, M Roayaei Ardakani, M Montazeri, N Kouhi Esfahani, N Asadimanesh, S Khalili and Z Payandeh. Dual roles of extracellular vesicles in acute lymphoblastic leukemia: Implications for disease progression and theranostic strategies. Medical Oncology 2024; 42(1), 11.
[4] H Kantarjian and E Jabbour. Adult acute lymphoblastic leukemia: 2025 update on diagnosis, therapy, and monitoring. American Journal of Hematology 2025; 100(7), 1205-1231.
[5] QL Ekpa, PC Akahara, AM Anderson, OO Adekoya, OO Ajayi, PO Alabi, OE Okobi, O Jaiyeola and MS Ekanem. A review of Acute Lymphocytic Leukemia (ALL) in the pediatric population: Evaluating current trends and changes in guidelines in the past decade. Cureus 2023; 15(12), e49930.
[6] R Salvaris and PL Fedele. Targeted therapy in acute lymphoblastic leukaemia. Journal of Personalized Medicine 2021; 11(8), 715.
[7] J Liu, D Tran, L Xue, BJ Wiley, C Vlasschaert, CJ Watson, HAJ MacGregor, X Zong, ICC Chan, I Das, MM Uddin, A Niroula, G Griffin, BL Ebert, T Mack, Y Pershad, B Sharber, M Berger, A Zehir, …, KL Bolton. Germline genetic variation impacts clonal hematopoiesis landscape and progression to malignancy. Nature Genetics 2025; 57(8), 1872-1880.
[8] F El Chaer, M Keng and KK Ballen. MLL-rearranged acute lymphoblastic leukemia. Current Hematologic Malignancy Reports 2020; 15(2), 83-89.
[9] N Aziz, YH Hong, HG Kim, JH Kim and JY Cho. Tumor-suppressive functions of protein lysine methyltransferases. Experimental & Molecular Medicine 2023; 55(12), 2475-2497.
[10] L Reed, J Abraham, S Patel and SS Dhar. Epigenetic modifiers: Exploring the roles of histone methyltransferases and demethylases in cancer and neurodegeneration. Biology 2024; 13(12), 1008.
[11] M Górecki, I Kozioł, A Kopystecka, J Budzyńska, J Zawitkowska and M Lejman. Updates in KMT2A Gene rearrangement in pediatric acute lymphoblastic leukemia. Biomedicines 2023; 11(3), 821.
[12] C Meyer, P Larghero, BALopes, T Burmeister, D Gröger, R Sutton, NC Venn, G Cazzaniga, LC Abascal, G Tsaur, L Fechina, M Emerenciano, MS Pombo-de-Oliveira, T Lund-Aho, T Lundán, M Montonen, V Juvonen, J Zuna, J Trka, …, R Marschalek. The KMT2A recombinome of acute leukemias in 2023. Leukemia 2023; 37(5), 988-1005.
[13] L Oksa, A Mäkinen, A Nikkilä, N Hyvärinen, S Laukkanen, A Rokka, P Haapaniemi, M Seki, J Takita, O Kauko, M Heinäniemi and O Lohi. Arginine methyltransferase PRMT7 deregulates expression of RUNX1 target genes in T-cell acute lymphoblastic leukemia. Cancers 2022; 14(9), 2169.
[14] A Montanaro, S Kitara, E Cerretani, M Marchesini, C Rompietti, L Pagliaro, A Gherli, A Su, ML Minchillo, M Caputi, R Fioretzaki, B Lorusso, L Ross, G Alexe, E Masselli, M Marozzi, FMA Rizzi, RL Starza, C Mecucci, …, G Roti. Identification of an Epi-metabolic dependency on EHMT2/G9a in T-cell acute lymphoblastic leukemia. Cell Death & Disease 2022; 13(6), 551
[15] M Khodadoust and O Silva. Hepatosplenic T-cell lymphoma with STAT5B and SETD2 mutations recurring as cells with NK-cell immunophenotype. Blood, The Journal of the American Society of Hematology 2023; 141(5), 555.
[16] J Li, J Hlavka-Zhang, JH Shrimp, C Piper, D Dupéré-Richér, JS Roth, D Jing, HLC Román, C Troche, A Swaroop, M Kulis, JA Oyer, CM Will, M Shen, A Riva, RL Bennett, AA Ferrando, MD Hall, RB Lock and JD Licht. PRC2 inhibitors overcome glucocorticoid resistance driven by NSD2 mutation in pediatric acute lymphoblastic leukemia. Cancer Discovery 2022; 12(1), 186-203.
[17] DY Nie, JR Tabor, J Li, M Kutera, J St-Germain, RP Hanley, E Wolf, E Paulakonis, TMG Kenney, S Duan, S Shrestha, DDG Owens, MER Maitland, A Pon, M Szewczyk, AJ Lamberto, M Menes, F Li, LZ Penn, …., CH Arrowsmith. Recruitment of FBXO22 for targeted degradation of NSD2. Nature Chemical Biology 2024; 20(12), 1597-1607.
[18] SW Brady, KG Roberts, Z Gu, L Shi, S Pounds, D Pei, C Cheng, Y Dai, M Devidas, C Qu, AN Hill, D Payne-Turner, X Ma, I Iacobucci, P Baviskar, L Wei, S Arunachalam, K Hagiwara, Y Liu, …, CG Mullighan. The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 2022; 54(9), 1376-1389.
[19] M Mounir, M Lucchetta, TC Silva, C Olsen, G Bontempi, X Chen, H Noushmehr, A Colaprico and E Papaleo. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Computational Biology 2019; 15(3), e1006701.
[20] D Long, Y Xue, X Yu, X Qin, J Chen, J Luo, K Ma, L Wei and X Li. Integrative multi-omics analysis reveals the molecular characteristics, tumor microenvironment, and clinical significance of ubiquitination mechanisms in lung adenocarcinoma. International Journal of Molecular Sciences 2025; 26(13), 6501.
[21] E Capriotti, P Fariselli and R Casadio. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Research 2005; 33(S2), W306-W310.
[22] DK Sarker, P Ray, FBA Salam and SJ Uddin. Exploring the impact of deleterious missense nonsynonymous single nucleotide polymorphisms in the DRD4 gene using computational approaches. Scientific Reports 2025; 15(1), 3150.
[23] MT Terry. A package for survival analysis in R. R package version 3.8-3, Available at https://CRAN.R-project.org/package=survival, accessed June 2025.
[24] Y Chen, L Chen, ATL Lun, PL Baldoni and GK Smyth. edgeR v4: Powerful differential analysis of sequencing data with expanded functionality and improved support for small counts and larger datasets. Nucleic Acids Research 2025; 53(2), gkaf018.
[25] C W Law, Y Chen, W Shi and GK Smyth. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology 2014; 15(2), R29.
[26] M Shahjaman, MMH Mollah, MR Rahman, SMS Islam and MNH Mollah. Robust identification of differentially expressed genes from RNA-seq data. Genomics 2020; 112(2), 2000-2010.
[27] Z Gu. Complex heatmap visualization. iMeta 2022; 1(3), e43.
[28] S Xu, E Hu, Y Cai, Z Xie, X Luo, L Zhan, W Tang, Q Wang, B Liu, R Wang, W Xie, T Wu, L Xie and G Yu. Using clusterProfiler to characterize multiomics data. Nature Protocols 2024; 19(11), 3292-3320.
[29] J Bendl, J Stourac, O Salanda, A Pavelka, ED Wieben, J Zendulka, J Brezovsky and J Damborsky. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Computational Biology 2014; 10(1), e1003440.
[30] R Reshmi and D Kaur. In silico analysis of non-synonymous single nucleotide polymorphisms of human ABCD1 gene associated with adrenoleukodystrophy. Egyptian Journal of Medical Human Genetics 2025; 26(1), 144.
[31] EA Stone and A Sidow. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome research 2005; 15(7), 978-986.
[32] E Capriotti and P Fariselli. PhD-SNPg: Updating a webserver and lightweight tool for scoring nucleotide variants. Nucleic Acids Research 2023; 51(W1), W451-W458.
[33] I Adzhubei, DM Jordan and SR Sunyaev. Predicting functional effect of human missense mutations using PolyPhen-2. Current Protocols in Human Genetics 2013; 76(1), 7-20.
[34] PC Ng and S Henikoff. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research 2003; 31(13), 3812-3814.
[35] NL Sim, P Kumar, J Hu, S Henikoff, G Schneider and PC Ng. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Research 2012; 40(W1), W452-W457.
[36] Y Bromberg, G Yachdav and B Rost. SNAP predicts effect of mutations on protein function. Bioinformatics 2008; 24(20), 2397-2398.
[37] H Tang and PD Thomas. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016; 32(14), 2230-2232.
[38] J Ma, Y Zhang, J Li, Y Dang and D Hu. Regulation of histone H3K27 methylation in inflammation and cancer. Molecular Biomedicine 2025; 6(1), 14.
[39] H Chen, S Wang, R Dong, P Yu, T Li, L Hu, M Wang, Z Qian, H Zhou, X Yue, L Wang and H Xiao. KDM6A deficiency induces myeloid bias and promotes CMML-like disease through JAK/STAT3 activation by repressing SOCS3. Advanced Science 2025; 12(21), 2413091.
[40] H Chen, H Huang and H Xiao. Kdm6a modulates hematopoiesis and leukemogenesis via demethylase-dependent epigenetic programming. Blood 2023; 142(S1), 1372-1372.
[41] S Chen, D Liu, B Chen, Z Li, B Chang, C Xu, N Li, C Feng, X Hu, W Wang, Y Zhang, Y Xie, Q Huang, Y Wang, S D Nimer, S Chen, Z Chen, L Wang and X Sun. Catalytic activity of Setd2 is essential for embryonic development in mice: Establishment of a mouse model harboring patient-derived Setd2 mutation. Frontiers of Medicine 2024; 18(5), 831-849.
[42] RM Chavez, DR Powell, K Lakhani, J Attelah, E Flynt, T Connolly, M Hamilton, G Mulligan, D Auclair, J Keats, PM Vertino, LH Boise, S Lonial, KN Conneely and BG Barwick. Ectopic NSD2 remodels H3K36me2 and DNA methylation to promote oncogenic gene expression in multiple myeloma. Blood 2024; 144(S1), 1359.
[43] PSY Chong, JY Chooi, JSL Lim, SHM Toh, TZ Tan and WJ Chng. SMARCA2 is a novel interactor of NSD2 and regulates prometastatic PTP4A3 through chromatin remodeling in t(4;14) multiple myeloma. Cancer Research 2021; 81(9), 2332-2344.
[44] K Wang, F Zhan, X Yang, M Jiao, P Wang, H Zhang, W Shang, J Deng and L Wang. KMT2D: A key emerging epigenetic regulator in head and neck diseases and tumors. Life Sciences 2025; 12(21), 2413091.