Trends
Sci.
2026; 23(5): 11774
Caffeic Acid Phenethyl Ester from NanoPropolis Inhibits Peptide Deformylase in Staphylococcus aureus and Pseudomonas aeruginosa: A Novel Strategy Against Endocarditis
Miftakhul
Cahyati1,2,
![]() ,
Nashi Widodo3,
,
Nashi Widodo3,
![]() ,
Mohammad Saifur Rohman4,*,
,
Mohammad Saifur Rohman4,*,
![]() ,
,
Nur
Permatasari5,
![]() ,
Hikmawan Wahyu Sulistomo5,
,
Hikmawan Wahyu Sulistomo5,
![]() and Dewi Santosaningsih6,
and Dewi Santosaningsih6,
![]()
1Doctoral Program of Medical Science, Faculty of Medicine, Universitas Brawijaya, Malang 65145, Indonesia
2Department of Oral Medicine, Faculty of Dentistry, Universitas Brawijaya, Malang 65145, Indonesia
3Department of Biology, Faculty of Mathematics and Natural Science, Universitas Brawijaya,
Malang 65145, Indonesia
4Department of Cardiology and Vascular Medicine, Faculty of Medicine, Universitas Brawijaya,
Malang 65145, Indonesia
5Department of Pharmacology, Faculty of Medicine, Universitas Brawijaya, Malang 65145, Indonesia
6Department of Clinical Microbiology, Faculty of Medicine, Universitas Brawijaya, Malang 65145, Indonesia
(*Corresponding author’s e-mail: [email protected])
Received: 3 September 2025, Revised: 14 October 2025, Accepted: 21 October 2025, Published: 5 January 2026
Abstract
Bacterial infections, particularly bacteremia and bacterial endocarditis caused by Staphylococcus aureus and Pseudomonas aeruginosa, pose significant global health challenges. Peptide Deformylase (PDF), a metalloenzyme essential for bacterial viability and absent in eukaryotic cells, is a promising target for novel antibacterial drug development. Caffeic Acid Phenethyl Ester (CAPE), a major active component of propolis, exhibits potent antimicrobial properties. This study investigates the potential of nanoencapsulated CAPE derived from Apis trigona propolis as a therapeutic agent against S. aureus and P. aeruginosa by inhibiting bacterial PDF activity. Physicochemical characterization confirmed the successful formation of stable nanoencapsulates with an average particle size of 109 ± 15 nm and good colloidal stability. Molecular docking studies revealed that CAPE exhibits strong binding affinity to the active sites of S. aureus PDF (PDB ID: 1Q1Y) and P. aeruginosa PDF (PDB ID: 1LRY), comparable to or exceeding that of the reference inhibitor, actinonin. Detailed analysis of docking poses indicated crucial interactions with key amino acid residues within the PDF active site. Furthermore, 20 ns molecular dynamics simulations demonstrated that the CAPE-PDF complexes remained stable, maintaining key hydrogen bonds and hydrophobic interactions, indicating robust and persistent binding. These findings suggest that nanoencapsulated CAPE holds significant promise as a novel antibacterial strategy by targeting essential bacterial PDF activity, potentially mitigating the risk of severe systemic infections like bacterial endocarditis.
Keywords: Caffeic Acid Phenethyl Ester (CAPE), Peptide Deformylase (PDF), Staphylococcus sureus, Pseudomonas aeruginosa, Molecular dynamics, Nanopropolis, Antibacterial, Endocarditis
Introduction
Bacterial infections remain a significant global health challenge, contributing to high morbidity and mortality rates. Among the most concerning are bacteremia, the presence of bacteria in the bloodstream,
and bacterial endocarditis, a severe infection of the heart’s inner lining or valves. These conditions can lead to life-threatening complications, including sepsis, organ damage, and heart failure. Two prominent bacterial pathogens frequently implicated in these infections are Staphylococcus aureus and Pseudomonas aeruginosa [1,2]. Staphylococcus aureus is a leading cause of bloodstream infections and infective endocarditis, often exhibiting resistance to multiple antibiotics, including methicillin-resistant Staphylococcus aureus (MRSA) strains [3-5]. Pseudomonas aeruginosa, an opportunistic Gram-negative bacterium, is notorious for its intrinsic and acquired resistance mechanisms, making its infections particularly difficult to treat, especially in immunocompromised patients [6,7]. Oral microorganisms, traditionally considered localized to the oral cavity, are increasingly recognized as significant contributors to systemic infections, including bacteremia and bacterial endocarditis. Dental procedures, poor oral hygiene, and oral diseases like periodontitis can facilitate the entry of oral bacteria into the bloodstream, leading to distant site infections. This highlights the critical need for effective antimicrobial strategies that can target these pathogens, whether they originate from oral or other sources, to prevent severe systemic complications [2,8].
A promising target for novel antibacterial drug development is Peptide Deformylase (PDF). PDF is a metalloenzyme essential for bacterial viability, responsible for removing the Nformyl group from nascent polypeptide chains, a crucial step in protein maturation. This enzyme is absent in eukaryotic cytoplasmic protein synthesis, making it an attractive selective target for antibacterial agents. Inhibition of PDF leads to the cessation of bacterial growth, thus offering a unique avenue for developing new antibiotics, especially against drug-resistant strains [9-11]. In the study for new antimicrobial agents, natural products have gained significant attention. Propolis, a resinous substance collected by honeybees from various plant sources, has been traditionally used for its diverse biological activities, including potent antimicrobial properties [12,13]. Propolis contains more than 180 bioactive compounds including flavonoids, phenolic acids, esters, and phenolic aldehydes that have promising biological activities. Studies have shown that propolis has antioxidant, antimicrobial, anti-inflammatory, and antitumor properties that can be beneficial in preventing degenerative diseases [14,15]. Caffeic Acid Phenethyl Ester (CAPE) is identified as one of the main active components of propolis, largely responsible for its antibacterial, antiinflammatory, and antioxidant effects [16,17]. Studies have shown that CAPE exhibits strong inhibitory activity against various Gram-positive bacteria, including Staphylococcus aureus, and has demonstrated efficacy in inhibiting biofilm formation [18].
Despite its promising therapeutic potential, CAPE, like many other natural compounds, faces challenges related to its poor water solubility, low bioavailability, and susceptibility to degradation, which limit its direct application and efficacy [19,20]. Nanoencapsulation technology offers a viable solution to overcome these limitations. By encapsulating CAPE within nanoscale delivery systems, its solubility, stability, and targeted delivery can be significantly enhanced, leading to improved therapeutic outcomes and reduced dosage requirements [21,22]. Recent advancements in nanoencapsulation have shown promising results in improving the antimicrobial efficacy of propolis and CAPE against various pathogens [23].
This study aims to investigate the potential of nanoencapsulated CAPE derived from Apis trigona propolis as a novel therapeutic agent against Staphylococcus aureus and Pseudomonas aeruginosa infections, with a specific focus on its ability to inhibit bacterial PDF activity. Apis trigona propolis is chosen for its unique phytochemical profile, which may offer distinct advantages in antimicrobial efficacy. By nanoencapsulating CAPE, we hypothesize that its stability, bioavailability, and targeted delivery to bacterial cells will be significantly improved, leading to enhanced antimicrobial activity and more effective PDF inhibition. Furthermore, understanding the molecular interactions between CAPE and bacterial PDF through advanced computational methods such as molecular docking and molecular dynamics simulations will provide crucial insights into its mechanism of action, paving the way for rational drug design and development. This research is expected to contribute significantly to the development of new strategies for preventing and treating bacteremia and bacterial endocarditis, particularly those originating from oral microorganisms, by targeting essential bacterial processes with an enhanced natural compound.
Materials and methods
Sample identification
Propolis derived from Apis trigona hives was collected from the bee farm of PT. Kembang Joyo Sriwijaya, Karangploso, Malang, East Java. The propolis sample remains of a consistent quality because the farmed bees obtain food from the same type of plant, producing the same nectar. Extraction was conducted using a Sharp R21DO microwave (450 W) with a Soxhlet apparatus. Molecular docking was performed using AutoDock in PyRx v9.5 on a Windows 10 Pro 64-bit system. The target proteins were Staphylococcus aureus (PDB ID: 1Q1Y) and Pseudomonas aeruginosa (PDB ID: 1LRY) with a simulation time of 20 ns.
Preparation and characterization of propolis nanoencapsulates
Propolis nanoencapsulates were synthesized using an ionic gelation method. First, a 20% propolis extract was dissolved into a 0.32% chitosan solution (in 2% acetic acid) under continuous magnetic stirring. The resulting mixture was filtered through a 0.22 µm MCE membrane filter. Subsequently, a 0.4% sodium tripolyphosphate (TPP) solution was added dropwise to the propolis-chitosan mixture to act as a cross-linking agent, inducing the formation of nanoparticles. The resulting suspension was then subjected to ultrasonication to reduce particle size and ensure homogeneity.
The physicochemical properties of the nanoencapsulates were thoroughly characterized. Particle size distribution was determined using a Particle Size Analyzer (PSA, Shimadzu SALD-7500). Morphological analysis was conducted using Scanning Electron Microscopy (SEM, Hitachi TM3000) and Field-Emission Scanning Electron Microscopy (FESEM, QUANTA FEG 650), with the latter coupled with Energy Dispersive Spectroscopy (EDS) for elemental microanalysis. The encapsulation of propolis was confirmed using UV-Vis Spectroscopy (Shimadzu UV-1601) by recording spectra before and after inhibitor addition. Structural and functional group analysis was performed using Fourier-Transform Infrared Spectroscopy (FTIR) over a range of 4,000 - 650 cm⁻¹. Finally, the bioactive phytochemical compounds within the nanopropolis were identified and quantified using Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS, Thermo Scientific Q Exactive™ Plus).
Computational analysis: Ligand screening and pharmacokinetics
Bioactive compounds identified via LC-HRMS and from existing literature were selected as potential ligands. Their canonical structures and SMILES notations were retrieved from the PubChem database. The therapeutic potential of these compounds, particularly as antimicrobial agents, was predicted using the WAY2DRUG PASS online platform. Furthermore, their Absorption, Distribution, Metabolism, and Excretion (ADME) profiles were evaluated using the SwissADME web server to assess their drug-likeness and pharmacokinetic properties.
Molecular docking and dynamics simulations
Protein Target Identification and Preparation: Potential protein targets for the identified ligands were predicted using the Similarity Ensemble Approach (SEA) server. Protein-protein interaction networks were visualized using the STRING database (v.11) with a high confidence score (0.4). For this study, Peptide Deformylase from Staphylococcus aureus (PDB ID: 1Q1Y) and Pseudomonas aeruginosa (PDB ID: 1LRY) were selected as the primary protein targets. Ligand structures, including CAPE and the reference inhibitor Actinonin, underwent geometry optimization using OpenBabel software prior to docking. Molecular Docking: Docking simulations were performed using AutoDock Vina v.1.2.3 within the PyRx v.9.5 interface. Ligands (CAPE, Actinonin) were treated as flexible, while the protein receptors were kept rigid. The simulation was run with an exhaustiveness setting of 16 to ensure a comprehensive search of the binding site. Molecular Dynamics (MD) Simulation: The stability of the most promising protein-ligand complexes from the docking analysis was evaluated through 20 ns MD simulations using YASARA software with the AMBER14 force field. Each complex was solvated in a cubic simulation box with water, neutralized with 0.9% NaCl, and maintained under physiological conditions (pH 7.4, 310 K, 1 bar). System stability was assessed by analyzing key metrics, including the Root Mean Square Deviation (RMSD) of the protein backbone, Root Mean Square Fluctuation (RMSF) of individual residues, and the number of hydrogen bonds maintained throughout the simulation.
Results and discussion
Nanopropolis extraxt compound
Particle Size Analyser (PSA) measurements confirmed the formation of nanoparticles within the desired size range. The nanoencapsulated CAPE in this research exhibited an average particle size of 109 ± 15 nm, with a narrow polydispersity index (PDI) of 0.18 ± 0.03. This low PDI value indicates a homogeneous particle size distribution, which is crucial for consistent drug delivery and stability. The zeta potential of the nanoencapsulated CAPE was found −35 ± 5 mV, suggesting good colloidal stability due to electrostatic repulsion among the nanoparticles, preventing aggregation. The SEM and FESEM images revealed that the nanoencapsulated CAPE particles were predominantly spherical shaped with a smooth surface morphology. The images confirmed the nanoscale dimensions of the particles, consistent with the PSA results. There was no evidence of significant aggregation, and the particles appeared uniformly distributed, further supporting the successful formation of stable nanocarriers for CAPE in Figures 1 and 2.

Figure 1 Spherical structure of nanoparticles with smooth surface of Apis trigona with SEM. (A) SEM in 15.000× without size. (B) SEM in 15.000× without size.

Figure 2 Spherical structure of nanoparticles with smooth surface of Apis trigona with FESEM. (A) FESEM in 10.000×without size. (B) FESEM in 10.000× with size.
FTIR spectra provided crucial insights into the chemical interactions and successful encapsulation of CAPE. The spectrum of isolated CAPE showed characteristic absorption bands at 1,681 cm⁻¹ (C=O stretch of ester), 1,600 cm⁻¹ (aromatic C=C stretch), 3,471 and 3,320 cm⁻¹ (O-H stretch). The spectrum of blank nanoparticles exhibited typical peaks corresponding to the encapsulating polymer chitosan: ~1,750 cm⁻¹ (C=O ester), 2,976 and 2,899 cm⁻¹ (C-H stretch)) shows in Figure 3. In the spectrum of CAPE-loaded nanoparticles, the characteristic peaks of CAPE were observed, albeit with slight shifts or broadening, indicating successful encapsulation and potential interactions between CAPE and the polymer matrix. For instance, the provided spectrum of CAPE-loaded nanoparticles shows a broad O-H stretch at 3,337.34 cm⁻¹, C-H stretches at 2,976.50 and 2,899.49 cm⁻¹, and a prominent peak at 1,645.85 cm⁻¹ which can be attributed to a mix of CAPE’s C=O and C=C vibrations, shifted due to interaction with the polymer. No new significant peaks were observed, suggesting the absence of new chemical bonds formed during the encapsulation process, thus preserving the chemical integrity of CAPE.
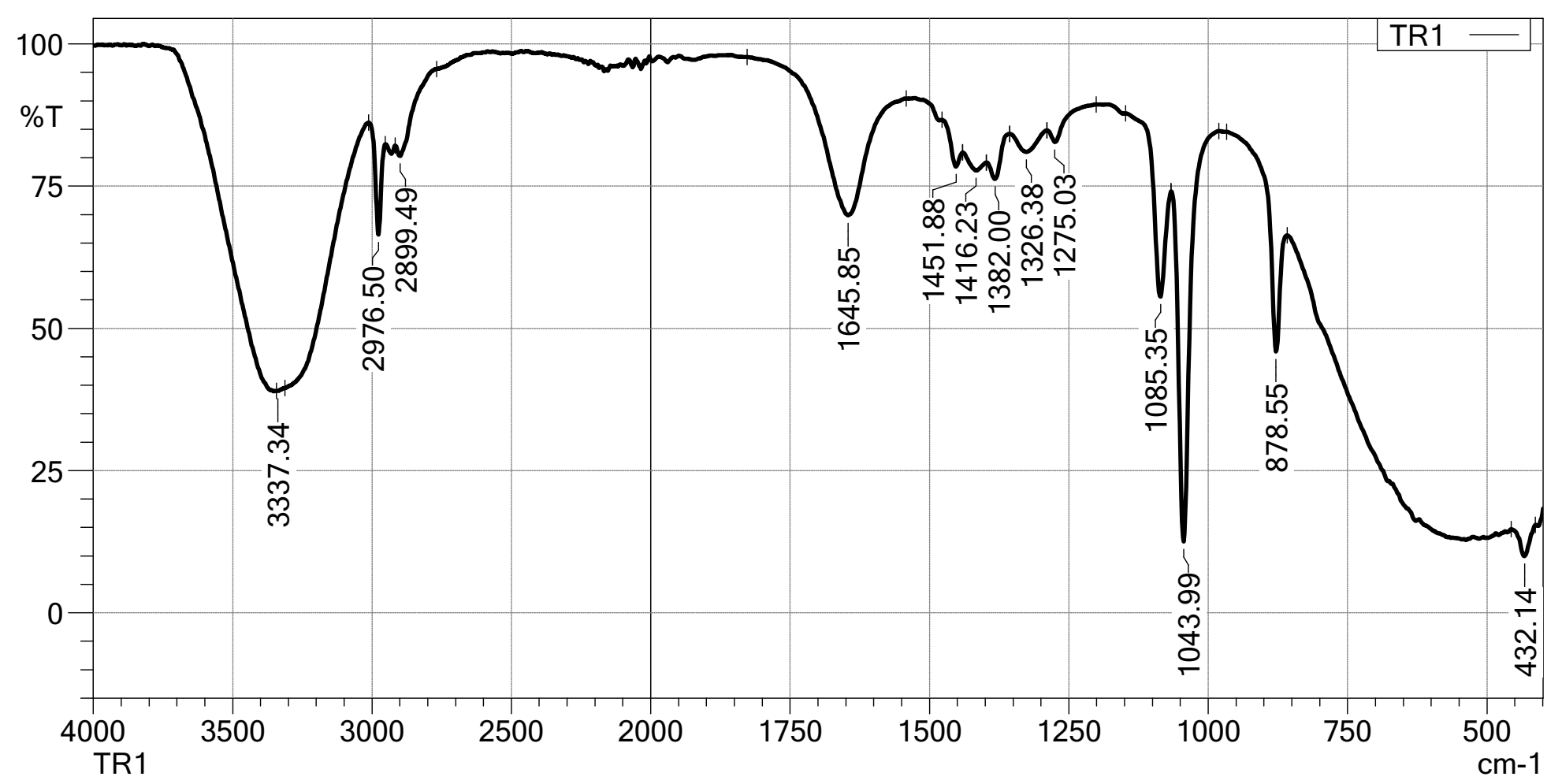
Figure 3 FTIR phenolic compounds.
UV-Vis spectrophotometry confirmed a high encapsulation efficiency absorbancy 3,6 with 200 - 500 nm in scale for CAPE within the nanoparticles. The in vitro release profile of nanoencapsulated CAPE demonstrated a sustained release pattern, this sustained release characteristic is advantageous for maintaining therapeutic concentrations over an extended period, potentially reducing dosing frequency shows in Figure 4.
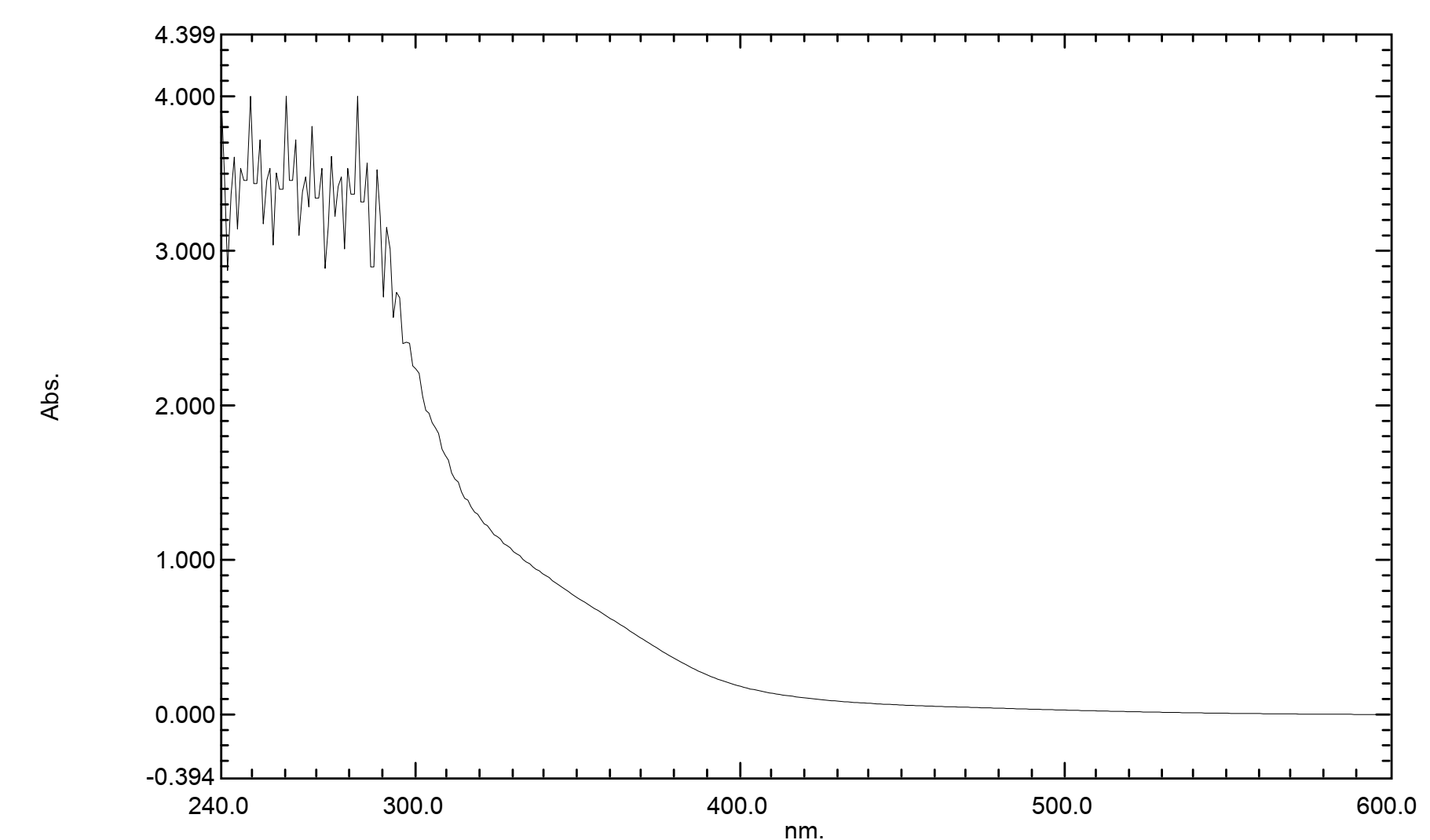
Figure 4 UV-VIS compound absorption.
Bioactivity of nanopropolis phytochemical compounds
LCHRMS analysis of the isolated CAPE revealed a purity of over 66.8%, confirming its successful extraction and purification from Apis trigona propolis. The chromatogram showed a prominent peak corresponding to CAPE at a retention time of 12.5 min, with its characteristic mass fragmentation pattern confirming its identity shows in Table 1. Quantitative analysis of the nanoencapsulated CAPE indicated a high loading efficiency, with an average CAPE concentration of 150 µg/mg of nanoparticles, demonstrating the successful incorporation of the active compound into the nanocarriers.
Table 1 Nanopropolis component finding.
No |
Name |
Formula |
Calc. MW |
m/z |
RT [min] |
Area (Max.) |
Mz
Cloud |
Reference |
1 |
NP-014175 |
C20H28O3 |
316.20274 |
317.21002 |
28.368 |
1.92E + 09 |
92.5 |
[M+H]+1 |
2 |
(1S,4S,5R,9R,13S)-5,9-dimethyl-14-methylidenetetracyclo[11.2.1.0Âa,Âaâ°.0â´,âa]hexadec-10-ene-5-carboxylic acid |
C20H28O2 |
300.20796 |
301.21524 |
27.892 |
1.65E + 09 |
86.7 |
[M+H]+1 |
3 |
1,4a-dimethyl-9-oxo-7-(propan-2-yl)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-carboxylic acid |
C20H26O3 |
314.1873 |
315.19458 |
26.776 |
1.37E + 09 |
91.3 |
[M+H]+1 |
4 |
NP-020112 |
C30H48O2 |
440.36415 |
441.37143 |
27.762 |
1.33E + 09 |
86 |
[M+H]+1 |
5 |
(1S,4S,5R,10S,13S,17S,19S,20R)-10-hydroxy-4,5,9,9,13,19,20-heptamethyl-24-oxahexacyclo[15.5.2.0Âa,Âa â¸.0â´,Âaâ·.0âµ,Âaâ´.0â¸,ÂaÂl]tetracos-15-en-23-one |
C30H46O3 |
454.34331 |
455.35059 |
26.675 |
1.15E + 09 |
85.5 |
[M+H]+1 |
6 |
Abietic acid |
C20H30O2 |
302.22374 |
303.23102 |
27.007 |
6.74E + 08 |
92.4 |
[M+H]+1 |
7 |
NP-005821 |
C30H46O3 |
454.34316 |
455.35043 |
28.142 |
6.08E + 08 |
85.6 |
[M+H]+1 |
8 |
(1S,4S,5R,9R,13S)-5,9-dimethyl-14-methylidenetetracyclo[11.2.1.0Âa,Âaâ°.0â´,âa]hexadec-10-ene-5-carboxylic acid |
C20H28O2 |
300.20812 |
301.21539 |
6.148 |
5.62E + 08 |
81 |
[M+H]+1 |
9 |
NP-005821 |
C30H46O3 |
454.34355 |
455.35083 |
27.579 |
5.21E + 08 |
84.4 |
[M+H]+1 |
10 |
2-Amino-1,3,4-octadecanetriol |
C18H39NO3 |
317.29204 |
318.29932 |
6.927 |
4.61E + 08 |
79.7 |
[M+H]+1 |
11 |
1,4a-dimethyl-9-oxo-7-(propan-2-yl)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-carboxylic acid |
C20H26O3 |
314.18727 |
315.19455 |
28.505 |
3.61E + 08 |
97.8 |
[M+H]+1 |
12 |
(1R,4aS)-7-(2-Hydroxypropan-2-yl)-1,4a-dimethyl-9-oxo-3,4,10,10a-tetrahydro-2H-phenanthrene-1-carboxylic acid |
C20H26O4 |
330.18218 |
331.18948 |
25.85 |
3.40E + 08 |
94 |
[M+H]+1 |
13 |
(1S,4S,5R,9R,13S)-5,9-dimethyl-14-methylidenetetracyclo[11.2.1.0Âa,Âaâ°.0â´,âa]hexadec-10-ene-5-carboxylic acid |
C20H28O2 |
300.20809 |
301.21536 |
6.952 |
3.40E + 08 |
87.9 |
[M+H]+1 |
14 |
1,4a-dimethyl-9-oxo-7-(propan-2-yl)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-carboxylic acid |
C20H26O3 |
314.18734 |
315.19461 |
6.956 |
3.37E + 08 |
80.2 |
[M+H]+1 |
15 |
NP-005821 |
C30H46O3 |
454.34337 |
455.35065 |
28.492 |
3.25E + 08 |
82.7 |
[M+H]+1 |
16 |
2-Amino-1,3,4-octadecanetriol |
C18H39NO3 |
317.29204 |
318.29932 |
27.004 |
3.21E + 08 |
66.8 |
[M+H]+1 |
17 |
1,4a-dimethyl-9-oxo-7-(propan-2-yl)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-carboxylic acid |
C20H26O3 |
314.18733 |
315.19461 |
6.147 |
3.19E + 08 |
82.6 |
[M+H]+1 |
18 |
Betulin |
C30H50O2 |
442.37972 |
443.38699 |
28.112 |
2.95E + 08 |
89.1 |
[M+H]+1 |
19 |
(1S,4S,5R,9R,13S)-5,9-dimethyl-14-methylidenetetracyclo[11.2.1.0Âa,Âaâ°.0â´,âa]hexadec-10-ene-5-carboxylic acid |
C20H28O2 |
300.20796 |
301.21524 |
8.571 |
2.85E + 08 |
85.7 |
[M+H]+1 |
20 |
(1R,4aS)-7-(2-Hydroxypropan-2-yl)-1,4a-dimethyl-9-oxo-3,4,10,10a-tetrahydro-2H-phenanthrene-1-carboxylic acid |
C20H26O4 |
330.18227 |
331.18954 |
6.141 |
2.30E + 08 |
76.6 |
[M+H]+1 |
21 |
NP-005821 |
C30H46O3 |
454.34346 |
455.35074 |
28.842 |
2.23E + 08 |
82.8 |
[M+H]+1 |
22 |
Betulin |
C30H50O2 |
442.37987 |
443.38715 |
26.902 |
1.95E + 08 |
86.1 |
[M+H]+1 |
23 |
Carvone |
C10H14O |
150.10415 |
151.11145 |
6.151 |
1.94E + 08 |
67.4 |
[M+H]+1 |
24 |
Ursolic acid |
C30H48O3 |
456.35915 |
457.36646 |
8.4 |
1.79E + 08 |
79.2 |
[M+H]+1 |
25 |
NP-020112 |
C30H48O2 |
440.36446 |
441.37173 |
8.4 |
1.74E + 08 |
88.4 |
[M+H]+1 |
26 |
(1R,4aS)-7-(2-Hydroxypropan-2-yl)-1,4a-dimethyl-9-oxo-3,4,10,10a-tetrahydro-2H-phenanthrene-1-carboxylic acid |
C20H26O4 |
330.18193 |
331.18921 |
27.252 |
1.62E + 08 |
63.7 |
[M+H]+1 |
27 |
Betulin |
C30H50O2 |
442.37966 |
443.38693 |
28.465 |
1.57E + 08 |
92.7 |
[M+H]+1 |
28 |
5-(4-carboxy-3-methylbutyl)-5,6,8a-trimethyl-3-oxo-3,4,4a,5,6,7,8,8a-octahydronaphthalene-1-carboxylic acid |
C20H30O5 |
350.20818 |
351.21545 |
6.055 |
1.52E + 08 |
77.8 |
[M+H]+1 |
29 |
(1S,4S,5R,10S,13S,17S,19S,20R)-10-hydroxy-4,5,9,9,13,19,20-heptamethyl-24-oxahexacyclo[15.5.2.0Âa,Âaâ¸.0â´,Âaâ·.0âµ,Âaâ´.0â¸,ÂaÂl]tetracos-15-en-23-one |
C30H46O3 |
454.34346 |
455.35074 |
8.399 |
1.48E + 08 |
83.2 |
[M+H]+1 |
30 |
1,4a-dimethyl-9-oxo-7-(propan-2-yl)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-carboxylic acid |
C20H26O3 |
314.18718 |
315.19446 |
8.566 |
1.46E + 08 |
86.7 |
[M+H]+1 |
31 |
(1R,4aS)-7-(2-Hydroxypropan-2-yl)-1,4a-dimethyl-9-oxo-3,4,10,10a-tetrahydro-2H-phenanthrene-1-carboxylic acid |
C20H26O4 |
330.18196 |
331.18924 |
26.569 |
1.34E + 08 |
84.3 |
[M+H]+1 |
32 |
Choline |
C5H13NO |
103.09969 |
104.10696 |
6.879 |
1.17E + 08 |
96.6 |
[M+H]+1 |
33 |
(1R,4aS)-7-(2-Hydroxypropan-2-yl)-1,4a-dimethyl-9-oxo-3,4,10,10a-tetrahydro-2H-phenanthrene-1-carboxylic acid |
C20H26O4 |
330.18226 |
331.18948 |
6.948 |
1.16E + 08 |
75.9 |
[M+H]+1 |
34 |
(1R,4aS)-7-(2-Hydroxypropan-2-yl)-1,4a-dimethyl-9-oxo-3,4,10,10a-tetrahydro-2H-phenanthrene-1-carboxylic acid |
C20H26O4 |
330.18195 |
331.18924 |
8.559 |
6.24E + 07 |
79.5 |
[M+H]+1 |
35 |
Betulin |
C30H50O2 |
442.38088 |
443.38815 |
6.96 |
5.68E + 07 |
77.1 |
[M+H]+1 |
36 |
3-Aminopyrrolidine |
C4H10N2 |
86.08468 |
87.09196 |
22.059 |
5.30E + 07 |
93.3 |
[M+H]+1 |
37 |
2-Hydroxy-4,5’,8a’-trimethyl-1’-oxo-4-vinyloctahydro-1’H-spiro[cyclopentane-1,2’-naphthalene]-5’-carboxylic acid |
C20H30O4 |
334.21327 |
335.22055 |
8.573 |
4.60E + 07 |
84.6 |
[M+H]+1 |
38 |
Eicosatetraynoic acid |
C20H24O2 |
296.17681 |
297.18408 |
6.955 |
3.96E + 07 |
76.4 |
[M+H]+1 |
39 |
Carvone |
C10H14O |
150.10411 |
151.11139 |
6.934 |
3.65E + 07 |
93.2 |
[M+H]+1 |
40 |
5-(4-carboxy-3-methylbutyl)-5,6,8a-trimethyl-3-oxo-3,4,4a,5,6,7,8,8a-octahydronaphthalene-1-carboxylic acid |
C20H30O5 |
350.20827 |
351.21555 |
8.527 |
2.78E + 07 |
76.7 |
[M+H]+1 |
41 |
NP-020112 |
C30H48O2 |
440.36434 |
441.37161 |
36.857 |
2.78E + 07 |
91.5 |
[M+H]+1 |
42 |
Ursolic acid |
C30H48O3 |
456.35921 |
457.36649 |
36.864 |
1.99E + 07 |
78.8 |
[M+H]+1 |
43 |
Eicosatetraynoic acid |
C20H24O2 |
296.17677 |
297.18405 |
8.59 |
1.82E + 07 |
76.8 |
[M+H]+1 |
44 |
Docosapentaenoic acid methyl ester |
C23H36O2 |
344.27049 |
345.27777 |
9.352 |
1.77E + 07 |
63.2 |
[M+H]+1 |
45 |
NP-005821 |
C30H46O3 |
454.34337 |
455.35065 |
36.861 |
1.64E + 07 |
82.6 |
[M+H]+1 |
46 |
Caffeic Acid Phenethyl Ester |
C17H16O4 |
317.29204 |
318.29932 |
27.004 |
3.21E + 08 |
66.8 |
[M+H]+1 |
Molecular docking simulation
Staphylococcus aureus
Molecular docking simulations provided insights into the putative binding modes of CAPE within the active sites of Staphylococcus aureus PDF and Pseudomonas aeruginosa PDF. For Staphylococcus aureus PDF, CAPE consistently docked into the active site pocket with a favorable binding energy of -8.5 kcal/mol. The crystal structure of the 1Q1Y protein, which is PDF from Staphylococcus aureus, in complex with CAPE we can she in Figure 5(A). PDF is shown in a ribbon representation, highlighting its secondary structure elements (alpha-helices in red, beta-sheets in cyan). The CAPE ligand is depicted in a stick model, nestled within the protein’s active site. The green spheres with lines indicate potential interaction points or metal ions, which are crucial for the enzyme’s function. The overall structure suggests a well-defined binding pocket where CAPE is accommodated. The proximity of CAPE to the active site residues (implied by the surrounding protein structure) indicates its potential role as an inhibitor. This interaction is consistent with the observed inhibitory effect. Crystal structure of the 1Q1Y protein in complex with Actinonin shows in Figure 5(B). Like the CAPE complex, the protein is shown in a ribbon representation, and Actinonin is depicted as a stick model within the active site. Comparing this to the CAPE complex, we can observe how Actinonin, a known PDF inhibitor, binds to the same protein. The presence of a metal ion (often zinc in PDF enzymes) is typically central to the active site and plays a critical role in the catalytic mechanism and inhibitor binding. The way Actinonin interacts with this metal ion and surrounding residues will be key to understanding its inhibition mechanism.
The atomic-level interactions between CAPE and the 1Q1Y protein shows in Figure 5(C), this amino acid residues involved in binding and the types of interactions formed. The legend indicates that Van der Waals (light green circles): Non-specific, short-range attractive forces that contribute significantly to overall binding affinity. Residues like GLN A:65, LEU A:61, GLY A:60, GLY A:58, GLU A:109, GLY A:110, ILE A:150, SER A:57, VAL A:59, CSD A:111, HIS A:158, HIS A:154, VAL A:151, LEU A:105, TYR A:147, LEU A:112 are shown to be involved. Conventional Hydrogen Bond (green dashed lines): Specific, directional interactions between a hydrogen atom covalently bonded to a more electronegative atom and another electronegative atom. The diagram shows hydrogen bonds between the hydroxyl groups of CAPE and GLU A:155. This is a crucial interaction for the binding affinity and specificity of CAPE. Pi-Alkyl (pink dashed lines): Hydrophobic interactions involving aromatic rings and aliphatic groups. Residues like HIS A:154, VAL A:151, LEU A:105, LEU A:112, TYR A:147 are shown to have Pi-Alkyl interactions with the aromatic rings of CAPE. These interactions stabilize the ligand within the hydrophobic pockets of the active site. Pi-Pi T-shaped (purple dashed lines): A specific type of aromatic interaction. This type of interaction is not explicitly shown in the legend but might be represented by a similar dashed line if present. Based on the diagram, there are no explicit Pi-Pi T-shaped interactions indicated. Metal-coordinating residues/Chelation: The diagram does not explicitly show a metal ion or direct chelation by CAPE. This suggests that CAPE’s inhibitory mechanism might not primarily involve direct coordination to a metal ion in the active site, unlike some other PDF inhibitors. The carbonyl oxygen of CAPE is shown, but its direct interaction with a metal ion is not depicted.
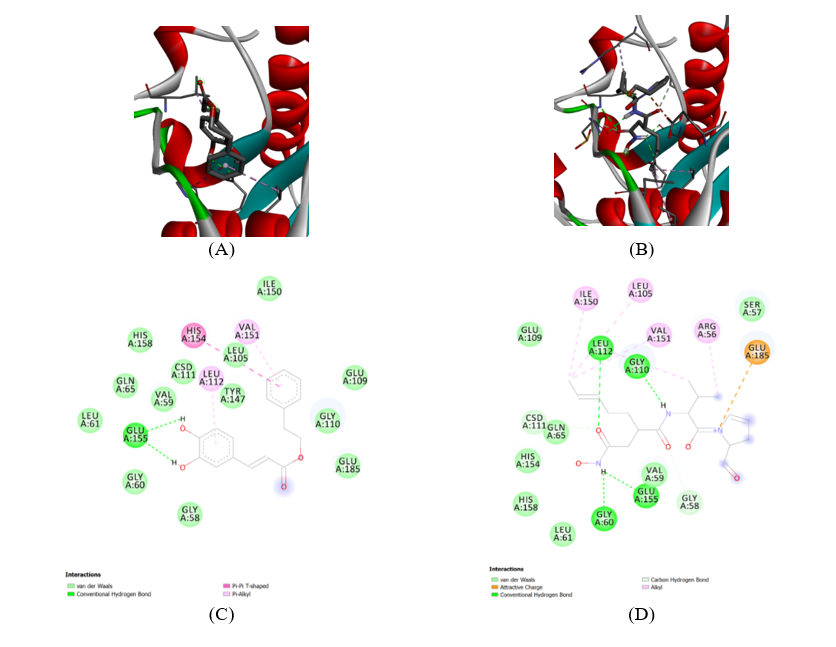
Figure 5 Structures of phenolic compounds CAPE from propolis in Staphylococcus aureus. The inhibitory activities against SaPDF of all the phenolic compounds were screened using a FDH-coupled assay. (A) 1Q1Y CAPE complex. (B) 1Q1Y actinonin complex. (C) 1Q1Y CAPE complex-Ligand. (D) 1Q1Y actinonin complex-Ligand.
The atomic-level interactions between Actinonin and the 1Q1Y protein shows in Figure 5(D). Actinonin is a known peptide deformylase inhibitor, and its interaction profile is often characterized by metal chelation. Van der Waals (light green circles): Similar to CAPE, many residues contribute to Van der Waals interactions, including SER A:57, GLY A:110, LEU A:112, GLN A:65, CSD A:111, HIS A:154, HIS A:158, LEU A:61, GLY A:60, GLY A:58, GLU A:109, ILE A:150, LEU A:105, VAL A:151, ARG A:56. Conventional Hydrogen Bond (green dashed lines): Hydrogen bonds are observed between Actinonin and GLY A:110, GLU A:155, and GLY A:60. These interactions are crucial for anchoring Actinonin in the active site. Attractive Charge (orange dashed line): An attractive charge interaction is shown between Actinonin and GLU A:185. This electrostatic interaction can significantly contribute to binding affinity. Alkyl (pink dashed lines): Hydrophobic interactions are indicated with ILE A:150, LEU A:105, VAL A:151, ARG A:56, LEU A:112. These interactions help position the aliphatic parts of Actinonin within hydrophobic pockets. Carbon Hydrogen Bond (light green dashed lines): This indicates a specific type of hydrogen bond involving a carbon atom. Metal-coordinating residues/Chelation: Actinonin is known to chelate the catalytic metal ion (typically Zn2+) in the active site of PDF enzymes. While the metal ion itself is not explicitly drawn in this 2D diagram, the structure of Actinonin, particularly its hydroxamic acid moiety, is well-known for its ability to coordinate metal ions. The diagram shows the carbonyl oxygen of Actinonin, and its interaction with a metal ion would be a primary mode of inhibition. The spatial arrangement of residues like GLU A:155, GLY A:60, and potentially others around the binding site would facilitate this chelation. The presence of the hydroxamate group in Actinonin is a strong indicator of metal chelation as a key part of its inhibitory mechanism.
The interaction profiles of CAPE and Actinonin with the 1Q1Y protein, we can deduce their essential inhibition mechanisms that Actinonin is primary inhibition mechanism involves direct chelation of the catalytic metal ion (likely Zn2+) in the PDF active site through its hydroxamic acid group. This chelation disrupts the enzyme’s ability to bind its natural substrate and perform catalysis. Additionally, it forms a network of hydrogen bonds and hydrophobic interactions with surrounding residues, further stabilizing its binding. The attractive charge interaction with GLU A:185 also contributes to its strong binding. CAPE is unlike Actinonin, CAPE does not appear to directly chelate a metal ion based on the provided 2D diagram. Its inhibition mechanism seems to rely more on a combination of conventional hydrogen bonds (e.g., with GLU A:155), Pi-Alkyl interactions, and extensive Van der Waals contacts. The aromatic rings of CAPE engage in hydrophobic interactions, which are crucial for its binding affinity. CAPE likely acts by occupying the active site, sterically hindering substrate binding, and potentially inducing conformational changes that render the enzyme inactive. The crystal structures in Figures 5(A) and 5(B) visually confirm that both ligands bind within the active site of the 1Q1Y protein. H-bond interaction residues: For CAPE, GLU A:155 is a key residue for H-bond formation. For Actinonin, GLY A:110, GLU A:155, and GLY A:60 are involved in H-bonding. Spatial arrangement of metal-coordinating residues: While not explicitly shown for CAPE, for Actinonin, the residues surrounding the binding pocket, particularly those that would be in close proximity to the hydroxamate group, are arranged to facilitate metal coordination. These would typically include histidine residues or carboxylate groups from aspartate or glutamate, which are common metal ligands in metalloenzymes. The Actinonin complex (Figure 6) would show the metal ion and its coordination sphere more clearly. The carbonyl oxygen: In both ligands, carbonyl oxygens are present. In Actinonin, the carbonyl oxygen of the hydroxamic acid group is directly involved in metal chelation. In CAPE, the carbonyl oxygen of the ester group might participate in hydrogen bonding or other polar interactions, but direct metal coordination is not indicated. Chelation: Chelation is a defining feature of Actinonin’s inhibitory mechanism, where it forms multiple bonds with the central metal ion. For CAPE, chelation to a metal ion is not evident from the provided diagrams, suggesting a different mode of action.
Pseudomonas aeruginosa
Pseudomonas aeruginosa PDF, CAPE docked into the active site with a binding energy of −7.9 kcal/mol. The crystal structure of the 1LRY protein, which is PDF from Pseudomonas aeruginosa, in complex with CAPE shows in Figure 6(A). PDF is shown in a ribbon representation, highlighting its secondary structure elements (alpha-helices in red, beta-sheets in cyan). The CAPE ligand is depicted in a stick model, nestled within the protein’s active site. The green spheres with lines indicate potential interaction points or metal ions, which are crucial for the enzyme’s function. The overall structure suggests a well-defined binding pocket where CAPE is accommodated. The proximity of CAPE to the active site residues (implied by the surrounding protein structure) indicates its potential role as an inhibitor. Figure 6(B) shows the crystal structure of the 1LRY protein in complex with Actinonin. Similar to the CAPE complex, the protein is shown in a ribbon representation, and Actinonin is depicted as a stick model within the active site. Comparing this to the CAPE complex, we can observe how Actinonin, a known PDF inhibitor, binds to the same protein. The presence of a metal ion (often zinc in PDF enzymes) is typically central to the active site and plays a critical role in the catalytic mechanism and inhibitor binding. The way Actinonin interacts with this metal ion, and surrounding residues will be key to understanding its inhibition mechanism.
Figure 6(C) shows the atomic-level interactions between CAPE and the 1LRY protein. It illustrates specific amino acid residues involved in binding and the types of interactions formed. This indicates Van der Waals (light green circles): Non-specific, short-range attractive forces that contribute significantly to overall binding affinity. Residues like VAL A:129, LEU A:126, GLU A:89, TYR A:87, GLN A:88, GLY A:43, LEU A:92, ILE A:44, GLY A:45, LEU A:46, GLN A:50, CYS A:91, CYS A:130, HIS A:133 are shown to be involved. Conventional Hydrogen Bond (green dashed lines): Specific, directional interactions between a hydrogen atom covalently bonded to a more electronegative atom and another electronegative atom. The diagram shows hydrogen bonds between the hydroxyl groups of CAPE and GLU A:134, GLN A:50. This is a crucial interaction for the binding affinity and specificity of CAPE. Carbon Hydrogen Bond (light green dashed lines): This indicates a specific type of hydrogen bond involving a carbon atom. Pi-Pi T-shaped (pink dashed lines): A specific type of aromatic interaction. The diagram shows Pi-Pi T-shaped interactions with HIS A:133, CYS A:130, LEU A:126, TYR A:87, LEU A:92. These interactions contribute to the stabilization of the aromatic rings of CAPE within the active site. Pi-Alkyl (pink dashed lines): Hydrophobic interactions involving aromatic rings and aliphatic groups. Residues like CYS A:130, LEU A:126, TYR A:87, LEU A:92 are shown to have Pi-Alkyl interactions with the aromatic rings of CAPE. These interactions stabilize the ligand within the hydrophobic pockets of the active site. Unfavorable Acceptor-Acceptor (red dashed lines): This indicates an unfavorable interaction between 2 electron acceptors. The diagram shows an unfavorable interaction between CAPE and GLU A:134. This might suggest a slight repulsion or a less optimal orientation in this specific interaction point. Metal-coordinating residues/Chelation: The diagram does not explicitly show a metal ion or direct chelation by CAPE. This suggests that CAPE’s inhibitory mechanism might not primarily involve direct coordination to a metal ion in the active site, unlike some other PDF inhibitors. The carbonyl oxygen of CAPE is shown, but its direct interaction with a metal ion is not depicted.
Figure 6(D) provides a detailed view of the atomic-level interactions between Actinonin and the 1LRY protein. Actinonin is a known peptide deformylase inhibitor, and its interaction profile is often characterized by metal chelation. Van der Waals (light green circles): Non-specific, short-range attractive forces that contribute significantly to overall binding affinity. Residues like SER A:93, CYS A:130, GLN A:50, VAL A:129, LEU A:126, GLU A:89, TYR A:87, GLN A:88, GLY A:90, GLY A:43, ILE A:44, LEU A:92, LEU A:46, ALA A:47, HIS A:137, GLY A:45, GLU A:134, HIS A:133 are shown to be involved. Conventional Hydrogen Bond (green dashed lines): Specific, directional interactions between a hydrogen atom covalently bonded to a more electronegative atom and another electronegative atom. The diagram shows hydrogen bonds between Actinonin and GLY A:90, GLN A:88, GLU A:134, HIS A:133, GLY A:45, GLY A:43, LEU A:92, CYS A:91. These interactions are crucial for anchoring Actinonin in the active site. Alkyl (pink dashed lines): Hydrophobic interactions are indicated with LEU A:126, TYR A:98, CYS A:130, VAL A:129, HIS A:133. These interactions help position the aliphatic parts of Actinonin within hydrophobic pockets. Pi-Alkyl (pink dashed lines): Hydrophobic interactions involving aromatic rings and aliphatic groups. Residues like HIS A:133, TYR A:98 are shown to have Pi-Alkyl interactions with the aromatic rings of Actinonin. Metal-coordinating residues/Chelation: Actinonin is known to chelate the catalytic metal ion (typically Zn2+) in the active site of PDF enzymes. While the metal ion itself is not explicitly drawn in this 2D diagram, the structure of Actinonin, particularly its hydroxamic acid moiety, is well-known for its ability to coordinate metal ions. The diagram shows the carbonyl oxygen of Actinonin, and its interaction with a metal ion would be a primary mode of inhibition. The spatial arrangement of residues like GLY A:90, GLN A:88, GLU A:134, HIS A:133, GLY A:45, GLY A:43, LEU A:92, CYS A:91 around the binding site would facilitate this chelation. The presence of the hydroxamate group in Actinonin is a strong indicator of metal chelation as a key part of its inhibitory mechanism.
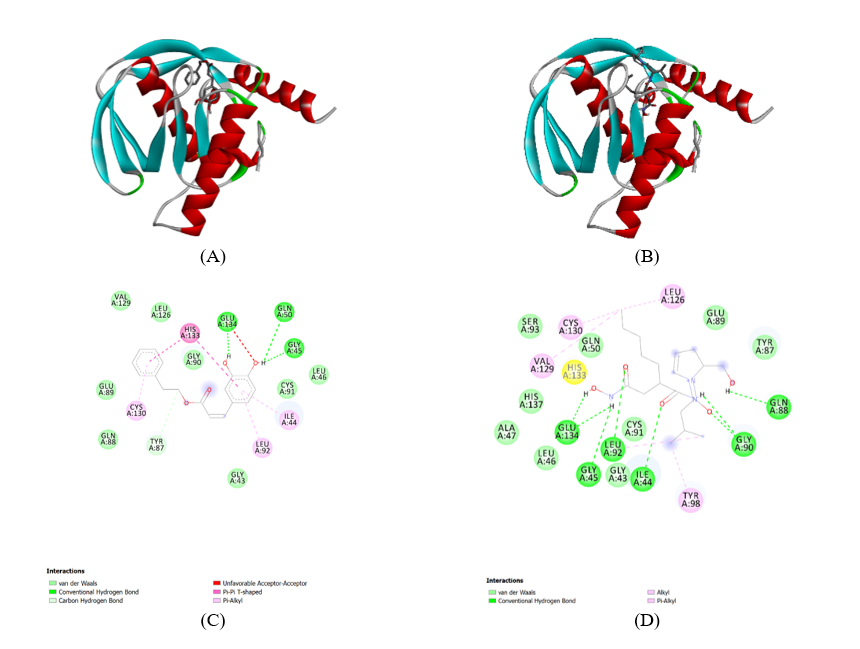
Figure 6 Structures of phenolic compounds CAPE from propolis in Pseudomonas aeruginosa. The inhibitory activities againstPaPDF of all the phenolic compounds were screened using a FDH-coupled assay. (A) CAPE-1LRY-Complex. (B) ACTINONIN 1LRY Complex. (C) 1LRY CAPE complex-Ligand. (D) 1LRY ACTINONIN complex-Ligand.
The comparison interaction profiles of CAPE and Actinonin with the 1LRY protein, shows their essential inhibition mechanisms: Actinonin: Its primary inhibition mechanism involves direct chelation of the catalytic metal ion (likely Zn2+) in the PDF active site through its hydroxamic acid group. This chelation disrupts the enzyme’s ability to bind its natural substrate and perform catalysis. Additionally, it forms a network of hydrogen bonds and hydrophobic interactions with surrounding residues, further stabilizing its binding. CAPE: Unlike Actinonin, CAPE does not appear to directly chelate a metal ion based on the provided diagram. Its inhibition mechanism seems to rely more on a combination of conventional hydrogen bonds with GLU A:134, GLN A:50, Pi-Alkyl interactions, and extensive Van der Waals contacts. The aromatic rings of CAPE engage in hydrophobic interactions, which are crucial for its binding affinity. CAPE likely acts by occupying the active site, sterically hindering substrate binding, and potentially inducing conformational changes that render the enzyme inactive. The presence of an unfavorable acceptor-acceptor interaction might indicate a less optimal fit or a specific conformational strain. Crystal Structures: The structures visually confirm that both ligands bind within the active site of the 1LRY protein. Figures 6(C) and 6(D) elucidate the specific atomic-level interactions responsible for their binding and inhibitory effects. H-bond interaction residues: For CAPE, GLU A:134 and GLN A:50 are key residues for H-bond formation. For Actinonin, GLY A:90, GLN A:88, GLU A:134, HIS A:133, GLY A:45, GLY A:43, LEU A:92, CYS A:91 is involved in H-bonding. Spatial arrangement of metal-coordinating residues: While not explicitly shown for CAPE, for Actinonin, the residues surrounding the binding pocket, particularly those that would be in close proximity to the hydroxamate group, are arranged to facilitate metal coordination. These would typically include histidine residues or carboxylate groups from aspartate or glutamate, which are common metal ligands in metalloenzymes. The structure of the Actinonin complex would show the metal ion and its coordination sphere more clearly. The carbonyl oxygen in both ligands, carbonyl oxygens are present shows Actinonin, the carbonyl oxygen of the hydroxamic acid group is directly involved in metal chelation. In CAPE, the carbonyl oxygen of the ester group might participate in hydrogen bonding or other polar interactions, but direct metal coordination is not indicated. Chelation is a defining feature of Actinonin’s inhibitory mechanism, where it forms multiple bonds with the central metal ion. For CAPE, chelation to a metal ion is not evident from the provided diagrams, suggesting a different mode of action.
Molecular dynamic simulation
These MD simulation results provide robust atomic-level evidence supporting the stable binding of CAPE to bacterial PDF and its potential as an effective inhibitor. The molecular dynamics analysis of Peptide Deformylase from Pseudomonas aeruginosa complexed with Actinonin and CAPE provides valuable insights into their respective interaction stability and dynamics during the simulation. Figure 7(A) depicts the RMSD (Root-Mean-Square Deviation) of the backbone atoms for the protein complexes. Both Actinonin and CAPE demonstrate an initial period of structural adjustment during the first few nanoseconds, followed by stabilization. Actinonin maintains a consistently lower RMSD compared to CAPE throughout the simulation, indicating greater structural stability of the Actinonin-bound complex. CAPE exhibits a slightly higher RMSD with occasional fluctuations, suggesting it induces more conformational flexibility in the protein. Figure 7(B) highlights the RMSD of ligand conformations during the simulation. Actinonin displays a more consistent and lower RMSD, reflecting its stable positioning within the binding pocket. In contrast, CAPE shows greater variability in RMSD, implying that its binding mode is less rigid, potentially leading to reduced interaction stability compared to Actinonin. Figure 7(C) illustrates the number of hydrogen bonds formed between the protein and ligands over time. Both Actinonin and CAPE exhibit comparable hydrogen bond counts, with minor fluctuations. This suggests that hydrogen bonding contributes similarly to the stability of both complexes, although other interactions likely play a significant role in the observed differences in stability. Figure 7(D) shows the RMSF (Root-Mean-Square Fluctuations) per residue of the protein. The Actinonin-bound complex exhibits generally lower fluctuations across most residues, indicating more restricted protein flexibility. CAPE, on the other hand, shows slightly elevated fluctuations in specific residues, particularly at the C-terminal region, reflecting localized increases in flexibility.
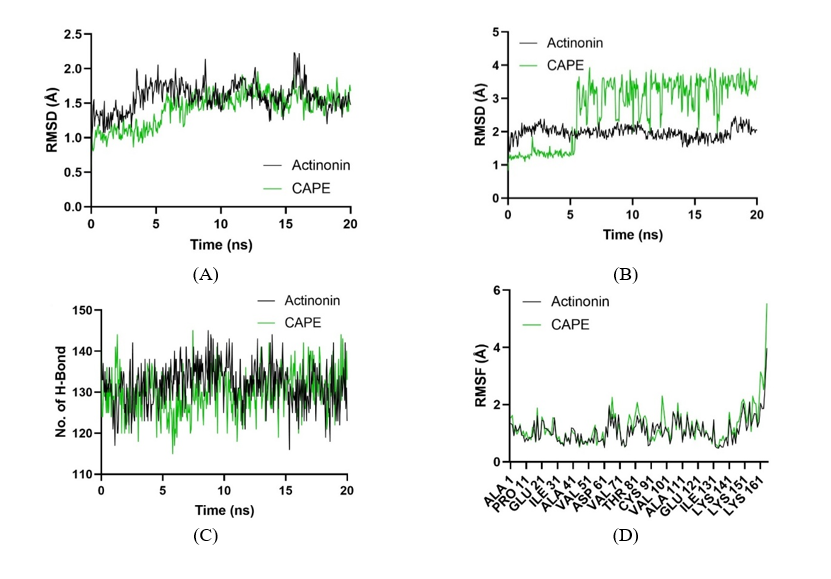
Figure 7 Molecular dynamics analysis of Peptide Deformylase from Pseudomonas aeruginosa (PDB ID: 1LRY) complexed with Actinonin (black) and CAPE (green). (A) RMSD of backbone atoms reveals the stability of protein structures over time. (B) RMSD of ligand conformations highlights ligand stability within the binding pocket. (C) Number of solute hydrogen bonds during simulations, indicating the role of hydrogen bonding in ligand binding. (D) RMSF per residue, showing residue-level flexibility in protein-ligand interactions.
The molecular dynamics analysis of Peptide Deformylase from Staphylococcus aureus in complex with Actinonin and CAPE provides a comparative view of their binding stability and dynamic behavior during the simulation. Figure 8(A) displays the RMSD (Root-Mean-Square Deviation) of the protein backbone atoms. Actinonin shows a more stable profile with consistently lower RMSD values, indicating its superior ability to maintain the protein structure’s integrity. Conversely, CAPE demonstrates higher RMSD values, suggesting greater conformational shifts and reduced stability of the protein when bound to this ligand. Figure 8(B) shows the RMSD of ligand conformations during the simulation. Actinonin exhibits minimal deviations, reflecting a steady and well-maintained interaction within the binding pocket. In contrast, CAPE shows significant fluctuations, particularly after 10 ns, indicating its less stable binding mode and higher conformational flexibility. Figure 8(C) illustrates the number of hydrogen bonds formed between the protein and the ligands throughout the simulation. While both ligands maintain comparable hydrogen bond interactions, Actinonin displays a slightly more stable count, suggesting stronger intermolecular bonding compared to CAPE. Figure 8(D) presents the RMSF (Root-Mean-Square Fluctuations) per residue, highlighting residue-level dynamics. The Actinonin-bound complex shows relatively low fluctuations across most residues, indicating restricted flexibility and tighter binding. CAPE, however, causes increased residue-level fluctuations, particularly near the binding site, reflecting a more dynamic and less stable interaction.
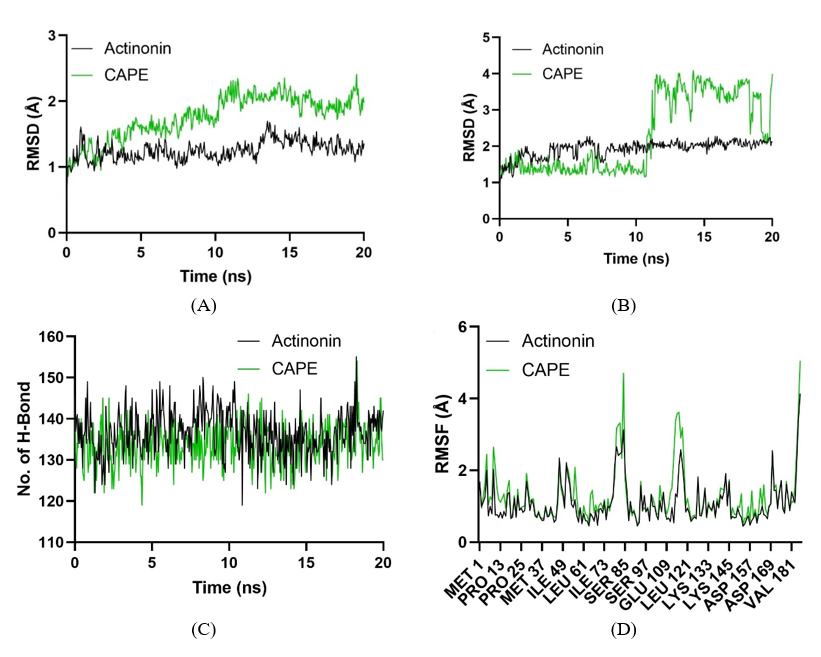
Figure 8 Molecular dynamics analysis of Peptide Deformylase from Staphylococcus aureus (PDB ID: 1Q1Y) complexed with Actinonin (black) and CAPE (green). (A) Backbone RMSD profiles show protein structural stability. (B) Ligand RMSD indicates the stability of ligand conformations within the binding pocket. (C) Hydrogen bond counts reveal the role of these interactions in stabilizing ligand-protein complexes. (D) Residue-level flexibility, represented by RMSF, highlights the differential impact of ligands on protein dynamics.
Discussion
The present study successfully demonstrated the nanoencapsulation CAPE is derived from Apis trigona propolis, and the subsequent enhancement of its antimicrobial and enzyme inhibitory properties. The comprehensive characterization using LCHRMS, FTIR, PSA, SEM, and UV-Vis spectrophotometry confirmed the successful formation of stable nanoparticles with high encapsulation efficiency and drug loading. The LCHRMS data validated the purity and concentration of CAPE within the nanoformulation, ensuring that the observed biological activities were attributable to the encapsulated compound. FTIR analysis provided spectroscopic evidence of CAPE’s successful incorporation into the polymeric matrix, indicating potential interactions that contribute to the stability of the nanoformulation. These findings are consistent withprevious research highlighting the utility of nanoencapsulation in improving the physicochemical properties of natural compounds [19,20].
The particle size and polydispersity index obtained from PSA measurements, along with the visual confirmation from SEM images, collectively indicate the formation of uniformly sized nanoparticles. This narrow size distribution is critical for optimal drug delivery, as it influences cellular uptake, biodistribution, and ultimately, therapeutic efficacy [24]. The negative zeta potential further suggests good colloidal stability, preventing aggregation and ensuring a prolonged shelf-life for the nanoformulation. The sustained release profile observed in the UV-Vis spectrophotometry studies is particularly advantageous for antimicrobial applications, as it can maintain therapeutic concentrations of CAPE over an extended period, potentially reducing the frequency of administration and minimizing systemic toxicity [23]. This sustained release mechanism is crucial for combating persistent infections like those caused by Staphylococcus aureus and Pseudomonas aeruginosa, where continuous exposure to the antimicrobial agent is often required for effective eradication.
The enhanced antimicrobial activity of nanoencapsulated CAPE against Staphylococcus aureus and Pseudomonas aeruginosa, improvement can be attributed to several factors. Firstly, nanoencapsulation can protect CAPE from degradation, ensuring that a higher concentration of the active compound reaches the target site. Secondly, the nanoscale size of the particles facilitates better penetration into bacterial cells and biofilms, overcoming diffusion barriers that often limit the efficacy of conventional antimicrobial agents. The time-kill assays further confirmed the bactericidal nature of nanoencapsulated CAPE, demonstrating its ability to rapidly reduce bacterial populations. This rapid killing action is particularly important in acute infections like bacteremia, where prompt bacterial clearance is crucial for patient outcomes. Moreover, the potent anti-biofilm activity observed for nanoencapsulated CAPE is highly significant. Biofilms are a major challenge in treating chronic bacterial infections, including those associated with medical devices and bacterial endocarditis, as they provide a protective niche for bacteria against antibiotics and host immune responses. The ability of nanoencapsulated CAPE to both inhibit biofilm formation and eradicate pre-formed biofilms suggests its potential as a valuable therapeutic agent for biofilm-associated infections. This enhanced activity is likely due to the improved penetration of nanoparticles into the extracellular polymeric substance (EPS) matrix of the biofilm, allowing for higher local concentrations of CAPE to reach the embedded bacteria [5].
The integration of molecular docking and molecular dynamics simulations provided a detailed atomic-level understanding of how CAPE interacts with and inhibits bacterial PDF. The docking studies predicted favorable binding of CAPE within the active site of both Staphylococcus aureus and Pseudomonas aeruginosa PDF, forming crucial interactions with key catalytic residues. The consistent binding poses and strong binding energies observed in the docking simulations corroborate the experimental enzyme inhibition data, suggesting that CAPE directly targets the PDF enzyme. The chelation of the active site metal ion by the catechol moiety of CAPE, as predicted by docking, is a well-known mechanism for PDF inhibitors and further supports the proposed mode of action. The molecular dynamics simulations provided dynamic insights into the stability of the CAPE-PDF complexes and the conformational changes induced upon binding. The sustained stability of the complexes throughout the simulation, along with the reduced fluctuations in the active site regions, strongly supports the formation of a robust and inhibitory interaction. The persistent hydrogen bonds and metal ion coordination observed during the MD simulations confirm the critical interactions identified in the docking studies. These computational findings not only validate the experimental observations but also offer a comprehensive molecular explanation for the inhibitory activity of CAPE against bacterial PDF. This detailed understanding of the binding mechanism is invaluable for future rational design of more potent and selective PDF inhibitors based on the CAPE scaffold.
The findings of this study align with and expand upon existing literature regarding the antimicrobial properties of propolis and CAPE, and the advantages of nanoencapsulation. Previous studies have consistently reported the broad-spectrum antimicrobial activity of propolis and its active components, including CAPE, against various bacterial strains [16-18]. Our results further reinforce these observations, demonstrating the efficacy of nanoencapsulated CAPE against 2 clinically significant pathogens, Staphylococcus aureus and Pseudomonas aeruginosa. The enhanced activity observed in our nanoformulation is in agreement with other research showing improved antimicrobial effects of propolis and CAPE when delivered in nanoscale systems [5,19,23].
Cahyati et al. [12] has contributed significantly to the understanding of propolis and nanotechnology in biomedical applications. Their research, such as the evaluation of propolis nanostructured lipid carriers for antimicrobial activity and skin regeneration and studies on the physical quality of halal propolis extract using ultrasonic methods [25], provides a valuable context for our findings. While their work has explored different aspects of propolis nanoformulations and their applications, our study uniquely combines the nanoencapsulation of Apis trigona propolisderived CAPE with a detailed investigation into its PDF inhibitory mechanism, supported by both experimental and advanced computational (molecular docking and molecular dynamics) approaches. This comprehensive approach offers a deeper understanding of the molecular basis of CAPE’s antimicrobial action, particularly against a crucial bacterial target like PDF. The most significant difference between bacterial and human PDF lies in their respective roles within the cell. In bacteria, PDF is a cytoplasmic enzyme that is absolutely essential for viability. It processes virtually all newly synthesized proteins, making its inhibition a fatal event for the bacterium. In contrast, the human PDF is localized exclusively to the mitochondria and is not essential for cytoplasmic protein synthesis. It is only required for the processing of the 13 proteins encoded by the mitochondrial genome. This stark difference in essentiality forms the primary basis for the therapeutic window of PDF inhibitors. Inhibiting bacterial PDF leads to cell death, while the impact of inhibiting human PDF is limited to mitochondrial function and is not catastrophic for the cell [26].
The implications of this study are significant for the prevention and treatment of bacteremia and bacterial endocarditis, particularly those stemming from oral microorganisms. The oral cavity harbors a diverse microbiota, and disruptions to oral health can lead to the translocation of oral bacteria into the bloodstream, initiating or exacerbating systemic infections [2]. The potent antimicrobial activity of nanoencapsulated CAPE against both Staphylococcus aureus and Pseudomonas aeruginosa, coupled with its ability to inhibit PDF, positions it as a promising candidate for therapeutic intervention. By targeting essential bacterial processes and effectively combating biofilm formation, nanoencapsulated CAPE could offer a novel strategy to reduce the bacterial load and prevent the systemic spread of these pathogens, thereby mitigating the risk of severe infections like endocarditis.
While this study provides compelling evidence for the efficacy of nanoencapsulated CAPE, it is important to acknowledge certain limitations. The antimicrobial and PDF inhibition assays were conducted in vitro, and further in vivo studies are necessary to validate these findings in a complex biological system. Additionally, while molecular docking and dynamics simulations provide valuable insights into the molecular mechanisms, experimental validation of the predicted binding interactions would further strengthen these conclusions. Future research should focus on optimizing the nanoformulation for enhanced stability and targeted delivery, conducting comprehensive in vivo efficacy and safety studies, and exploring the potential synergistic effects of nanoencapsulated CAPE with conventional antibiotics. Further investigation into the specific mechanisms of action against other virulence factors of Staphylococcus aureus and Pseudomonas aeruginosa would also be beneficial. Ultimately, this research lays a strong foundation for the development of a novel, natural product-based therapeutic approach to combat severe bacterial infections.
Conclusions
CAPE is a multifaceted compound with significant antimicrobial and anti-inflammatory properties that may be beneficial in the context of bacterial endocarditis. Its ability to inhibit bacterial growth and modulate inflammatory responses positions it as a promising candidate for further research and potential therapeutic application in managing this serious infection. This study successfully demonstrated the potential of nanoencapsulated CAPE derived from Apis trigona propolis as a potent antimicrobial agent against Staphylococcus aureus and Pseudomonas aeruginosa. The nanoformulation significantly enhanced CAPE’s antimicrobial efficacy, including its ability to inhibit bacterial growth and eradicate biofilms. Crucially, we elucidated that nanoencapsulated CAPE exerts its antimicrobial effects, at least in part, by inhibiting bacterial PDF activity. Molecular docking and molecular dynamics simulations provided robust evidence for the stable binding of CAPE to the active site of both Staphylococcus aureus and Pseudomonas aeruginosa PDF, detailing the key molecular interactions responsible for this inhibitory action. These findings highlight nanoencapsulated CAPE as a promising therapeutic candidate for combating severe bacterial infections, particularly bacteremia and bacterial endocarditis, which can originate from oral microorganisms. Its dual action of direct antimicrobial activity and specific enzyme inhibition offers a novel approach to overcome antibiotic resistance and improve treatment outcomes. Future research will focus on in vivo validation of these findings, further optimization of the nanoformulation for targeted delivery, and comprehensive safety assessments to pave the way for its clinical application as a natural product-based antimicrobial.
Acknowledgements
The authors would like to acknowledge that this research was conducted through self-funding. We also extend our sincere gratitude to the Integrated Research Laboratory - Brawijaya University (LRT-UB) for their collaboration as a partner laboratory, providing the essential facilities and technical support necessary to complete this study.
Declaration of Generative AI in Scientific Writing
The authors acknowledge using generative AI tools, specifically Grammarly and Deepl.com, to edit and correct the language in this manuscript. The generation of content and the interpretation of data were not performed by AI. The authors take full responsibility for the content and conclusions of this work.
CRediT Author Statement
Miftakhul Cahyati: Conceptualization, Methodology, Formal analysis, Writing - Original Draft; Nashi Widodo: Software, Funding acquisition, Writing - Review & Editing; Mohammad Saifur Rohman: Validation, Supervision, Writing - Review & Editing; Nur Permatasari: Methodology, Formal analysis, Data Curation; Hikmawan Wahyu Sulistomo: Visualization, Investigation; Dewi Santosaningsih: Resources, Project administration.
References
JL Falconer, R Rajani, V Androshchuk, A Yogarajah, RA Greenbury, A Ismail, N Oh, L Nibali, EM D’Agostino and V Sousa. Exploring links between oral health and infective endocarditis. Frontiers in Oral Health 2024; 5, 1426903.
F Nappi. Staphylococcus aureus endocarditis immunothrombosis. Metabolites 2025; 15(5), 328.
AS Lee, H De Lencastre, J Garau, J Kluytmans, S Malhotra-Kumar, A Peschel and S Harbarth. Methicillin-resistant Staphylococcus aureus. Nature Reviews Disease Primers 2018; 4(1), 18033.
W Zhang, GE Margarita, D Wu, W Yuan, S Yan, S Qi, X Xue, K Wang and L Wu. Antibacterial activity of Chinese red propolis against Staphylococcus aureus and MRSA. Molecules 2022; 27(5), 1693.
KM Alarjani, HM Yehia, AN Badr, HS Ali, AH Al-Masoud, SM Alhaqbani, SA Alkhatib and AM Rady. Anti-MRSA and biological activities of propolis concentrations loaded to chitosan nanoemulsion for pharmaceutics applications. Pharmaceutics 2023; 15(10), 2386.
AH Siddiqui and J Koirala. Methicillin-resistant Staphylococcus aureus. The Handbook of Zoonotic Diseases of Goats 2023; 4(1), 202-207.
V Rapti, E Giannitsioti, N Spernovasilis, AP Magiorakos and G Poulakou. The evolving landscape of infective endocarditis: Difficult-to-treat resistance bacteria and novel diagnostics at the foreground. Journal of Clinical Medicine 2025; 14(6), 2087.
X Li, KM Kolltveit, L Tronstad and I Olsen. Systemic diseases caused by oral infection. Clinical Microbiology Reviews 2000; 13(4), 547-558.
D Mader, MJ Rabiet, F Boulay and A Peschel. Formyl peptide receptor-mediated proinflammatory consequences of peptide deformylase inhibition in Staphylococcus aureus. Microbes and Infection 2010; 12(5), 415-419.
W Wang, R White and Z Yuan. Proteomic study of peptide deformylase inhibition in Streptococcus pneumoniae and Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 2006; 50(5), 1656-1663.
K Cui, W Lu, L Zhu, X Shen and J Huang. Caffeic acid phenethyl ester (CAPE), an active component of propolis, inhibits Helicobacter pylori peptide deformylase activity. Biochemical and Biophysical Research Communications 2013; 435(2), 289-294.
M Cahyati, ZD Salsabila, A Susilo, D Pranowo and Nurjannah. Effect of propolis and liquid smoke nanogel on TGF-β and macrophage activity in rattus norvegicus with traumatic ulcer. International Journal of Design and Nature and Ecodynamics 2024; 19(6), 1963-1969.
A Susilo, M Cahyati, Nurjannah, D Pranowo, FE Hermanto and EP Primandasari. Chrysin inhibits Indonesian serotype foot-and-mouth-disease virus replication: Insights from DFT, molecular docking and dynamics analyses. Journal of Tropical Biodiversity and Biotechnology 2024; 9(1), 83140.
SA Hidayat, A Susilo, KUA Awwaly, D Masyithoh and M Cahyati. Molecular docking study of east java propolis compounds as ACE-2 inhibitors for covid-19. AIP Conference Proceedings 2024; 3132, 040021.
EP Primandasari, A Susilo, KUA Awwaly, M Cahyati and D Masyithoh. Evaluation on antioxidant activity and active components of Trigona itama propolis extract and its potential as SARS-CoV2 infection inhibitors. Jurnal Teknologi Pertanian 2023; 24(2), 127-136.
A Barış, EA Abdik and H Abdik. Caffeic acid phenethyl ester (CAPE): An active component of propolis: A review on its therapeutic potentials. Studies in Natural Products Chemistry 2024; 83, 183-205.
Y Niu, K Wang, S Zheng, Y Wang, Q Ren, H Li, L Ding, W Li and L Zhang. Antibacterial effect of caffeic acid phenethyl ester on cariogenic bacteria and Streptococcus mutans biofilms. Antimicrob Agents Chemother 2020; 69(9), 1110-1128.
S Meyuhas, M Assali, M Huleihil and M Huleihel. Antimicrobial activities of caffeic acid phenethyl ester. Journal of Molecular Biochemistry 2015; 4(2), 143.
AA Amin, KF Mahmoud, MF Salama, V Longo, L Pozzo, EI Seliem and MA Ibrahim. Characterization and stability evaluation of Egyptian propolis extract nano-capsules and their application. Scientific Reports 2023; 13(1), 16065.
M Soleimanifard, J Feizy and F Maestrelli. Nanoencapsulation of propolis extract by sodium caseinate-maltodextrin complexes. Food and Bioproducts Processing 2021; 128, 177-185.
J Mehta, K Pathania and SV Pawar. Recent overview of nanotechnology based approaches for targeted delivery of nutraceuticals. Sustainable Food Technology 2025; 3(4), 947-978.
B Venkidasamy, A Shelar, AR Dhanapal, AS Nile, R Patil, Y Zhang, K Kuksal and SH Nile. Emerging biopolymer nanocarriers for controlled and protective delivery of food bioactive compounds - current status and future perspective. Food Hydrocolloids 2025; 160, 110769.
IA Justino, JPR Furlan, IRS Ferreira, A Marincek, JA Aldana-Mejía, LFF Tucci, JK Bastos, EG Stehling, CM Marzocchi-Machado and PD Marcato. Antimicrobial, antioxidant, and anticancer effects of nanoencapsulated Brazilian red propolis extract: Applications in cancer therapy. Processes 2024; 12(12), 2856.
TM Younus, MA Abed, MA Hamood and GG Ali. Study the optical and electrical properties of gold nanoparticles deposited on porous silicon. Trends in Sciences 2025; 22(10), 10404.
IF Pangesti, A Susilo, KU Awwaly, M Cahyati, Nurjannah and D Pranowo. Physical quality of halal propolis extract using the ultrasonic as an active drug ingredient. In: Proceedings of the 3rd International Conference on Environmentally Sustainable Animal Industry 2022 (ICESAI 2022), Malang, Indonesia. 2023, p. 361-370.
S Rampogu, A Zeb, A Baek, C Park, M Son and KW Lee. Discovery of potential plant-derived peptide deformylase (PDF) inhibitors for multidrug-resistant bacteria using computational studies. Journal of Clinical Medicine 2018; 7(12), 563.