Trends
Sci.
2026;
23(6):
12491
α-Glucosidase Inhibitory Constituents from the Fruits of Thai Helicteres isora L.
Phat Tan Mai1,2, Lien Thi My Do2, Thuy Thi Le Nguyen3, Rico Ramadhan4,5,
Suwimon Khwunsiriwong6, Thapakorn Chumphon7 and Jirapast Sichaem8,*
1Faculty of Medicine and Pharmacy, Vietnam College of Industry and Commerce,
Ho Chi Minh 760000, Vietnam
2Faculty of Natural Sciences Education, Sai Gon University, Ho Chi Minh 749000, Vietnam
3Department of Biotechnology, Ho Chi Minh City Open University, Ho Chi Minh 710000, Vietnam
4Division of Exploration and Synthesis of Bioactive Compounds (ESBC), CoE-University Research Center for Bio-Molecule Engineering (BIOME), Universitas Airlangga, Surabaya 60115, Indonesia
5Department of Chemistry, Faculty of Science and Technology, Universitas Airlangga, Surabaya 60115, Indonesia
6Department of Chemistry, Faculty of Science and Technology, Rajabhat Rajanagarindra University,
Chachoengsao 24000 Thailand
7Faculty of Agriculture at Kamphaeng Saen, Kasetsart University, Kamphaeng Saen Campus,
Nakhon Pathom 73140, Thailand
8Department of Chemistry, Faculty of Science and Technology, Thammasat University Lampang Campus,
Lampang 52190, Thailand
(*Corresponding author’s e-mail: [email protected])
Received: 1 November 2025, Revised: 4 December 2025, Accepted: 11 December 2025, Published: 20 February 2026
Abstract
Helicteres isora L. is a traditionally recognized medicinal plant in Asia, used for the treatment of gastrointestinal, metabolic, respiratory, and infectious diseases, with different parts exhibiting diverse pharmacological activities. In the present investigation, the fruits of H. isora L. were successfully investigated, leading to the isolation of β-sitosterol (1), betulinic acid (2), ursolic acid (3), 3 -hydroxyurs-11-ene-28(13)-lactone (4), ergosterol-5,8-peroxide (5), syringaresinol (6), β-sitosterol-3-O-β-D-glucoside (7), curcumin (8), demethoxycurcumin (9), and bisdemethoxycurcumin (10). The inhibitory effects of all isolated compounds on α-glucosidase and xanthine oxidase (XO) were assessed. Among them, compound 9 exhibited the most potent α-glucosidase inhibition (IC50 75.5 ± 0.06 µM), whereas compound 8 showed weak XO inhibition (IC50 95.0 ± 0.16 µM). In the molecular docking results, compounds 8 - 10 demonstrated strong α-glucosidase inhibition with conserved interactions at key residues (Asp203, Trp299, Trp406, Met444, Phe575, and Arg526), and their predicted physicochemical and pharmacokinetic properties support their potential as orally active, locally acting antidiabetic agents.
Keywords: Helicteres isora L., Phytochemicals, α-Glucosidase inhibition, Xanthine oxidase inhibition, Molecular docking, ADMET
Introduction
Diabetes mellitus (DM) is a long-term metabolic disease marked by dysregulated glucose homeostasis, often manifested as elevated postprandial blood glucose levels [1]. One promising therapeutic approach for managing postprandial hyperglycemia involves the inhibition of α-glucosidase, a key enzyme responsible
for carbohydrate hydrolysis. Inhibition of this enzyme delays glucose absorption in the intestine, thereby mitigating the rise in blood glucose levels following meals [2]. Natural products have attracted considerable attention as rich sources of α-glucosidase inhibitors, offering safer and more biocompatible alternatives to synthetic agents, which, despite their efficacy, are often associated with adverse effects [3]. Medicinal plants represent abundant reservoirs of structurally diverse bioactive constituents exhibiting broad pharmacological properties and relatively low toxicity. Traditionally, such plants have been widely employed in the management of chronic diseases, including diabetes, cancer, and cardiovascular disorders. Among these, Helicteres isora L. (Sterculiaceae), commonly referred to as the Indian screw tree, has long been used in traditional medicine systems throughout tropical and subtropical regions of Asia, particularly in India, Sri Lanka, and Thailand [4]. Its fruits are widely utilized in folk medicine for gastrointestinal ailments, diabetes, respiratory conditions, and microbial infections, while the roots, bark, and fruits have been reported to possess hypoglycemic, hypolipidemic, hepatoprotective, cardiotonic, and wound-healing activities [5]. These effects are attributed to a variety of secondary metabolites, including flavonoids, phenolics, tannins, alkaloids, glycosides, sterols, and terpenoids, some of which exhibit antioxidant, anti-inflammatory, antidiabetic, antimicrobial, and anticancer properties [6-8]. As part of our ongoing investigation into secondary metabolites and their α-glucosidase inhibitory potential from Thai medicinal plants [9,10], we focused on the fruits of H. isora L. and successfully isolated 10 compounds (1 - 10) (Figure 1). These compounds were then evaluated for their α-glucosidase inhibitory activity to assess their potential as natural antidiabetic agents, while their xanthine oxidase (XO) inhibitory activity was also examined.
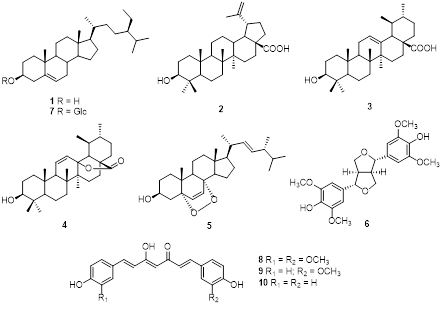
Figure 1 Chemical structures of compounds 1 - 10.
Materials and methods
General experimental procedures
Chromatographic separations were performed using silica gel 60 (0.040 - 0.063 mm, Merck, Darmstadt, Germany) and Sephadex LH-20 (GE Healthcare, Uppsala, Sweden). Fractions were monitored by thin-layer chromatography (TLC) on silica gel 60 F254 plates (Merck, Darmstadt, Germany). and visualized by spraying with 10% H2SO4 followed by heating. The structures of the isolated compounds were elucidated using NMR spectroscopy (Bruker AvanceNEO 600 MHz, Bruker Corporation, MA, USA) and electrospray ionization mass spectrometry (ESI-QTOF, AB Sciex, MA, USA). Absorbance was measured using an Agilent BioTek Epoch 2 microplate spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). α-Glucosidase (EC 3.2.1.20) from Saccharomyces cerevisiae (750 UN), p-nitrophenyl α-D-glucopyranoside (pNPG), and acarbose were purchased from Sigma-Aldrich (St. Louis, MO, USA). Xanthine oxidase (EC 1.17.3.2) from bovine milk (25 UN), xanthine, and allopurinol were also obtained from Sigma-Aldrich.
Plant material
Helicteres isora L. fruits were collected in Lampang Province, Thailand, in August 2024. The scientific name was identified by Asst. Prof. Dr. Kanit Wangwasit, Department of Biology, Faculty of Science, Mahasarakham University, Thailand, where the voucher specimen (K. Wangwasit 240811-1) has been deposited. The collection was conducted in accordance with all relevant local laws and ethical guidelines.
Extraction and isolation
The dried fruits of H. isora L. (15.0 kg) were macerated with 95% ethanol at room temperature (100 L×3). Removal of the solvent under reduced pressure gave a crude ethanol extract (650.0 g), which was partitioned with n-hexane and EtOAc to yield the n-hexane (145.0 g), EtOAc (230.0 g), and aqueous fractions. The EtOAc fraction was subjected to silica gel column chromatography (CC) using gradients of n-hexane:EtOAc (9:1 to 0:10, v/v) and EtOAc:MeOH (9:1 to 0:10, v/v), and TLC analysis allowed combining the fractions to afford 9 major fractions (EA.1 - EA.9). Fraction EA.2 (20.1 g) was further separated by silica gel CC (n-hexane:EtOAc, 8:2 to 0:10, v/v) to give 6 subfractions (EA.2.1 - EA.2.6). Subfraction EA.2.3 (3.7 g) was purified by silica gel CC (n-hexane: CHCl3, 8:2, v/v) and Sephadex LH-20 CC (CHCl3:MeOH, 1:4, v/v) to afford compounds 1 (35.5 mg) (Rf 0.50, n-hexane: CHCl3, 8:2, v/v) and 5 (6.5 mg) (Rf 0.40, n-hexane:CHCl3, 8:2, v/v). Fraction EA.3 (17.8 g) was fractionated into 4 subfractions (EA.3.1 - EA.3.4) by silica gel CC (n-hexane:EtOAc, 8:2 to 0:10, v/v). Subfraction EA.3.3 (3.7 g) was purified by silica gel CC (n-hexane:CHCl3, 7:3, v/v) and Sephadex LH-20 CC (CHCl3:MeOH, 1:4, v/v) to give compounds 2 (7.2 mg) (Rf 0.45, n-hexane:CHCl3, 7:3, v/v) and 3 (9.5 mg) (Rf 0.40, n-hexane:CHCl3, 7:3, v/v), while subfraction EA.3.4 (2.5 g) was similarly purified to afford compounds 4 (5.0 mg) (Rf 0.45, n-hexane:CHCl3, 7:3, v/v) and 6 (5.0 mg) (Rf 0.40, n-hexane:CHCl3, 7:3, v/v). Fraction EA.6 (19.0 g) was separated into 6 subfractions (EA.6.1 - EA.6.6) by silica gel CC (n-hexane:EtOAc, 7:3 to 0:10, v/v). Subfraction EA.6.3 (7.6 g) was purified with CHCl3:MeOH (98:2, v/v) followed by Sephadex LH-20 CC to yield compounds 8 (150.0 mg) (Rf 0.55, CHCl3:MeOH, 98:2, v/v), 9 (130.0 mg) (Rf 0.50, CHCl3:MeOH, 98:2, v/v), and 10 (150.0 mg) (Rf 0.50, CHCl3:MeOH, 98:2, v/v). Fraction EA.7 (18.2 g) was fractionated into 5 subfractions (EA.7.1 - EA.7.5) by silica gel CC (n-hexane:EtOAc 6:4 to 0:10, v/v; EtOAc:MeOH 9:1 to 0:10, v/v). Compound 7 (110.0 mg) (Rf 0.4, n-hexane:EtOAc, 1:1, v/v) was obtained from EA.7.3 (5.0 g) by silica gel CC (n-hexane:EtOAc, 1:1, v/v) (Scheme 1).
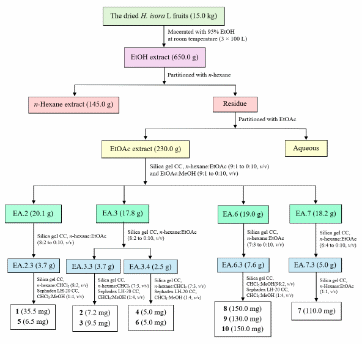
Scheme 1 Extraction and isolation procedures of compounds 1 - 10.
α-Glucosidase inhibition assay
The α-glucosidase inhibitory activity was evaluated using a previously reported method [10] with minor modifications. The assay was based on the enzymatic hydrolysis of p-nitrophenyl α-D-glucopyranoside (pNPG) by α-glucosidase. Samples were dissolved in DMSO at various concentrations (5 - 200 μg/mL), and the final DMSO content in the reaction mixture was maintained at 3%. The reaction mixture contained 60 μL of sodium phosphate buffer (100 mM, pH 6.8), 20 μL of sample solution, and 20 μL of α-glucosidase (0.3 IU/mL). The mixture was incubated in a 96-well plate at 37 °C for 10 min. The reaction was initiated by adding 100 μL of pNPG solution (200 μM), followed by an additional 10-min incubation at 37 °C, and then terminated with 100 μL of NaOH (50 mM). The release of p-nitrophenol (pNP) was measured at 405 nm. Percentage inhibition was calculated using the equation [(A0 − A1)/A0]×100, where A0 is the absorbance of the control and A1 is the absorbance in the presence of the sample. Negative controls without samples were run in parallel, and acarbose (50 - 150 μg/mL) was used as the positive control. IC50 values were determined by regression analysis from plots of percentage inhibition versus sample concentration.
Xanthine oxidase inhibition assay
The xanthine oxidase (XO) inhibitory activity was determined using xanthine as the substrate, following a previously described method [10]. In brief, 100 μL of xanthine oxidase solution (0.03 U/mL) in phosphate buffer (50 mM, pH 7.5) was mixed with 50 μL of the sample solution (5 - 200 μg/mL) and incubated in 96-well plates at 37 °C for 30 min. The reaction was initiated by adding 80 μL of xanthine solution (100 μM) in phosphate buffer and further incubated at 37 °C for 15 min. The reaction was then terminated by adding 30 μL of HCl solution (50 mM). Buffer and allopurinol (1 - 10 μg/mL) were used as the vehicle and positive control, respectively. Xanthine oxidase inhibitory activity was measured at 295 nm and expressed as percentage inhibition using the equation: [(A0 – A1)/A0]×100, where A0 is the absorbance of the control and A1 is the absorbance in the presence of the sample. Negative controls without samples were performed in parallel. The IC50 values for each sample were calculated by regression analysis from plots of percentage inhibition versus sample concentration.
In silico prediction of binding modes, physicochemical and pharmacokinetic properties of bioactive compounds
For molecular interaction analysis, the isolated active compounds were subjected to the molecular docking method to predict their binding affinity and mode with the target enzyme. Based on the superior inhibitory activity observed in the enzyme inhibition assays, the study focused on the α-glucosidase as the primary target. The structure of human intestinal α-glucosidase was retrieved from the RCSB PDB (PDB ID: 2QMJ) [11], while the xanthine oxidase structure was excluded from this subsequent computational analysis. Ligands and other small molecules associated with the enzyme structures were removed. No modeling or filling-in of missing residues was required before docking, as the structure was complete in the binding region. Protonation state corresponds to pH 7, which reflects the physiological condition in the human jejunum where α-glucosidase is most active. The structures of the active compounds were taken from PubChem database [12]. The docking process was run using AutoDock4 [13]. The center of the grid box was set at −21.572, −6.239, and −5.354, which corresponds to the center of the ligand (acarbose) in the reference structure. The size of the grid box was 60 60 60 points. The docking simulation used 200 runs of the Genetic Algorithm (GA runs) and the number of energy evaluations (ga_num_evals) was 25,000,000. All other AD4 parameters were left by default. Kollman charges were assigned to the protein, and the ligands prior to docking. 3D visualization was carried out using UCSF Chimera [14] while 2D interaction diagram was carried out using LigPlot+ [15]. Furthermore, physicochemical and pharmacokinetic properties which are major contributors to drug development were evaluated using ADMETLab platform [16].
Statistical analysis
The IC50 values were calculated from the slopes of the dose-response curves using Microsoft Excel. All results are presented as mean ± standard deviation (SD) from triplicate determinations. Differences among test samples were analyzed by 1-way analysis of variance (ANOVA), followed by Duncan’s multiple range test, with p < 0.05 considered statistically significant.
Results and discussion
Phytochemical identification
Chromatographic separation and spectroscopic analysis of H. isora L. fruits enabled the isolation and characterization of 10 compounds, β-sitosterol (1), betulinic acid (2), ursolic acid (3), 3 -hydroxyurs-11-ene-28(13)-lactone (4), ergosterol-5,8-peroxide (5), syringaresinol (6), β-sitosterol-3-O-β-D-glucoside (7), curcumin (8), demethoxycurcumin (9), and bisdemethoxycurcumin (10). The structures of these compounds were interpreted using extensive spectroscopic data in comparison with values reported in the literature.
Compound 1 was obtained as a white needle-like crystal. Its 1H NMR spectrum exhibited an olefinic proton signal at δH 5.26 (1H, m, H-6) and a carbinolic proton signal at δH 3.27 (1H, m, H-3). The 13C NMR spectrum indicated the presence of 29 carbon atoms, with resonances at δC 141.1 and 119.9 corresponding to the C=C bond between C-5 and C-6. Based on the NMR spectroscopic data (Tables 1 and 2) and comparison with previously reported data [17], compound 1 was identified as β-sitosterol, a phytosterol commonly found in plants.
Compound 2 appeared as a white solid. Its 1H NMR spectrum exhibited 2 exo-methylene proton resonances at δH 4.69 (1H, s, H-29b) and 4.56 (1H, s, H-29a), 6 methyl protons, and 6 methine protons. The 13C NMR spectrum displayed 30 carbon signals, including sp2 carbons at δC 150.3 (C-20) and 109.6 (C-29), an oxygenated carbon at δC 76.7 (C-3), and a carboxylic carbonyl carbon at δC 177.2 (C-28), along with 6 methyl and 5 quaternary carbons. From the observed NMR spectroscopic data (Tables 1 and 2) and comparison with previously published data [18], compound 2 was identified as a lupeol-type triterpenoid, betulinic acid.
Compound 3 was isolated as a white solid. Its 1H NMR spectrum displayed a signal at δH 5.13 (1H, s, H-12), and the 13C NMR spectrum revealed 30 carbon resonances, including a carboxylic carbonyl at δC 178.3 (C-28), 2 olefinic carbons at δC 124.6 (C-12) and 138.2 (C-13), and an oxygenated methine at δC 76.8 (C-3), along with 9 methylene, 7 methyl, 7 methine, and 7 quaternary carbons. Based on the NMR spectroscopic evidence (Tables 1 and 2) and comparison with previously reported literature data [19], compound 3 was characterized as ursolic acid, a pentacyclic triterpenoid of the ursane type.
Compound 4 was afforded as a white powder. The molecular formula, C30H46O3, was supported by electrospray ionization mass spectrum (ESIMS) [m/z 455.28 [M+H]+ (calcd. for [C30H46O3+H]+, 455.35). Its 1H NMR spectrum indicated an oxygenated methine at δH 3.21 (1H, dd, J = 13.8, 6.0 Hz), olefinic protons at δH 5.53 (1H, dd, J = 12.6, 3.6 Hz, H-11) and 5.96 (1H, d, J = 12.6 Hz, H-12), and 6 methyl signals. The 13C NMR spectrum exhibited 30 carbon signals, including 7 methyl, 1 oxygenated methine, 2 olefinic, 1 ester carbonyl, 8 methylene, 5 methine, and 6 quaternary carbons. On the basis of the NMR (Tables 1 and 2) and ESIMS spectroscopic data, and in comparison, with previously published values [19], compound 4 was identified as an ursane-type triterpenoid, 3β-hydroxyurs-11-ene-28(13)-lactone.
Compound 5 appeared as a white solid. The 1H and 13C NMR spectra displayed olefinic signals at δH 6.42 (1H, d, J = 8.5 Hz)/δC 130.6 (C-7) and 6.21 (1H, d, J = 8.5 Hz)/δC 136.0 (C-6) (Table 1), together with quaternary oxygenated carbons at δC 81.9 (C-5) and δC 78.9 (C-8), indicating a peroxide bridge between C-5 and C-8 (Table 2). The coupling constant (J6,7 = 8.5 Hz) confirmed the cis configuration of H-6 and H-7. The C-3 hydroxyl group was assigned a β-orientation, while the C-5/C-8 peroxide bridge was α-oriented. The remaining stereocenters were consistent with an ergostane-type skeleton. From the NMR spectroscopic data (Tables 1 and 2), together with comparison to earlier literature data [20], compound 5 was determined to be ergosterol-5,8-peroxide.
Compound 6 was isolated as a white waxy solid. Its 1H NMR spectrum showed a hydroxyl proton at δH 8.24 (1H, s, 4-OH), aromatic protons at δH 6.60 (4H, s, H-2,6,2′,6′), and 4 methoxy groups at δH 3.77 (12H, s, 3,5,3′,5′-OCH3). These spectral features are consistent with a symmetrical 3,7-dioxabicyclo[3.3.0]octane-type lignan skeleton. Regarding stereochemistry, bicyclooctane-type lignans are known to possess cis-configured protons, as supported by the observed coupling constants. Comparison of its NMR spectroscopic data (Table 3) with literature values [21] confirmed the identity of 6 as syringaresinol.
Compound 7 was obtained as a yellow solid. Its 1H and 13C NMR spectra were similar to those of compound 1, except for additional signals of 5 methine protons (δH 4.22 - 2.88) and 1 methylene group (δH 3.64 and 3.41). The 13C NMR spectrum also showed 6 carbons corresponding to a sugar moiety (δC 100.8, 76.9, 76.8, 73.4, 70.1, and 61.1), with an anomeric carbon at δC 100.8 (C-1′). According to the NMR spectroscopic data (Tables 1 and 2) and comparison with literature data [22], compound 7 was characterized as β-sitosterol-3-O-β-D-glucoside (daucosterol).
Compound 8 appeared as a light-yellow powder. Its molecular formula was confirmed as C21H20O6 by the ESIMS spectrum, based on the sodiated molecular ion peak at m/z 391.04 [M+Na]⁺ (calcd. for [C21H20O6+Na]+, 391.04). The 1H NMR spectrum displayed 2 trans-olefinic proton resonances at δH 7.53 (2H, d, J = 15.6 Hz, H-4,4′) and 6.74 (2H, d, J = 15.6 Hz, H-3,3′), along with 1 olefinic proton at δH 6.06 (1H, s, H-1). Two sets of AMX spin systems characteristic of 1,3,4-trisubstituted benzene rings were observed at δH 9.66 (2H, s, H-8,8′), 7.31 (2H, d, J = 1.8 Hz, H-6,6′), 7.15 (2H, dd, J = 8.4, 1.8 Hz, H-10,10′), and 6.82 (2H, d, J = 7.8 Hz, H-9,9′), together with 2 methoxy groups at δH 3.83 (6H, s, 8,8′-OCH3). The 13C NMR spectrum exhibited 21 carbon signals, including 1 carbonyl carbon at δC 183.2 (C-2′), 12 aromatic carbons (δC 149.4 - 111.4), 6 olefinic carbons (δC 183.2 - 100.8), and 2 methoxy carbons at δC 55.7 (Table 4). Analysis of the NMR (Table 4) and ESIMS spectroscopic features, together with comparison to previously published data [23], indicated that compound 8 is the acyclic diarylheptanoid curcumin.
Compound 9 was isolated as a light-yellow powder. Its 1H and 13C NMR spectra data closely resembled those of compound 8, except for the absence of a methoxy group at C-7, which was replaced by a proton. The aromatic region exhibited an AA′XX′ spin system at δH 7.56 (2H, d, J = 8.4, 1.6 Hz, H-6,10) and 6.81 (2H, dd, J = 8.4, 1.6 Hz, H-7,9), corresponding to carbon signals at δC 159.8 (C-8), 130.3 (C-6,10), 126.4 (C-5), and 115.7 (C-7,9), indicative of a 1,4-disubstituted benzene ring. From the NMR spectroscopic data (Table 4) and comparison with previously reported literature [23], compound 9 was assigned as demethoxycurcumin
Compound 10 was afforded as a light-yellow powder. Its molecular formula was assigned as C19H16O4 based on the ESIMS spectrum, which showed a sodiated molecular ion peak at m/z 331.16 [M+Na]⁺ (calcd. for [C19H16O4+Na]+, 331.09). Comparison of the 1H and 13C NMR spectral data with those of compounds 8 and 9 revealed that compound 10 is also an acyclic diarylheptanoid. The 1H NMR spectrum exhibited AA′XX′ spin systems at δH 7.57 (4H, d, J = 9.0 Hz, H-6,6′,10,10′) and 6.82 (4H, d, J = 8.4 Hz, H-7,7′,9,9′), indicative of 2 para-disubstituted aromatic rings. Corresponding 13C NMR signals were observed at δC 159.8 (C-8,8′), 130.3 (C-6,6′,10,10′), 125.9 (C-5,5′), and 115.9 (C-7,7′,9,9′). The observed NMR spectroscopic features (Table 4), in comparison with previously reported data [23], led to the assignment of compound 10 as bisdemethoxycurcumin.
Table 1 1H NMR data (600 MHz) of compounds 1 - 5 and 7 in DMSO-d6 (δ in ppm, J in Hz).
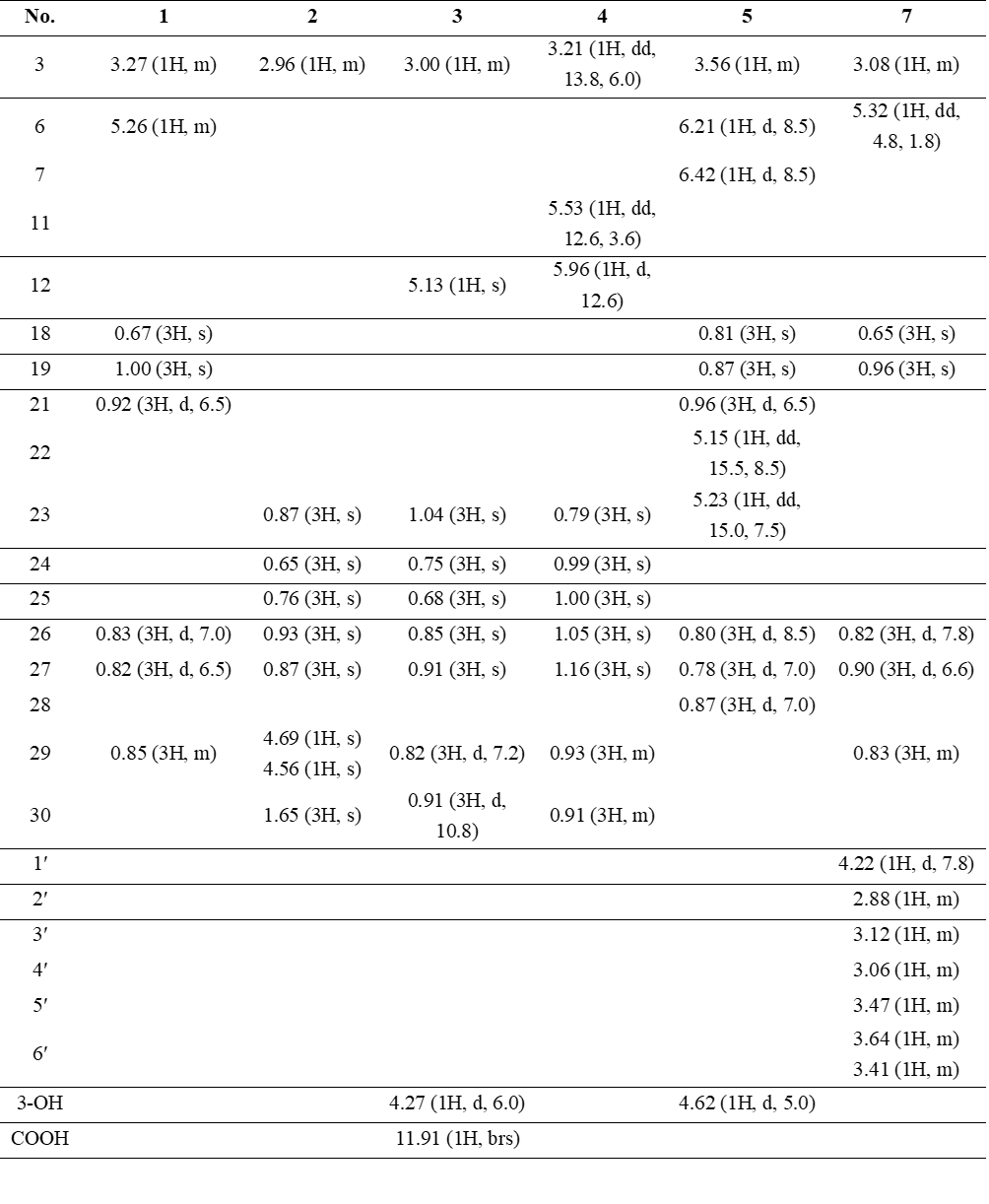
Table 2 13C NMR data (150 MHz) of compounds 1 - 5 and 7 in DMSO-d6 (δ in ppm).
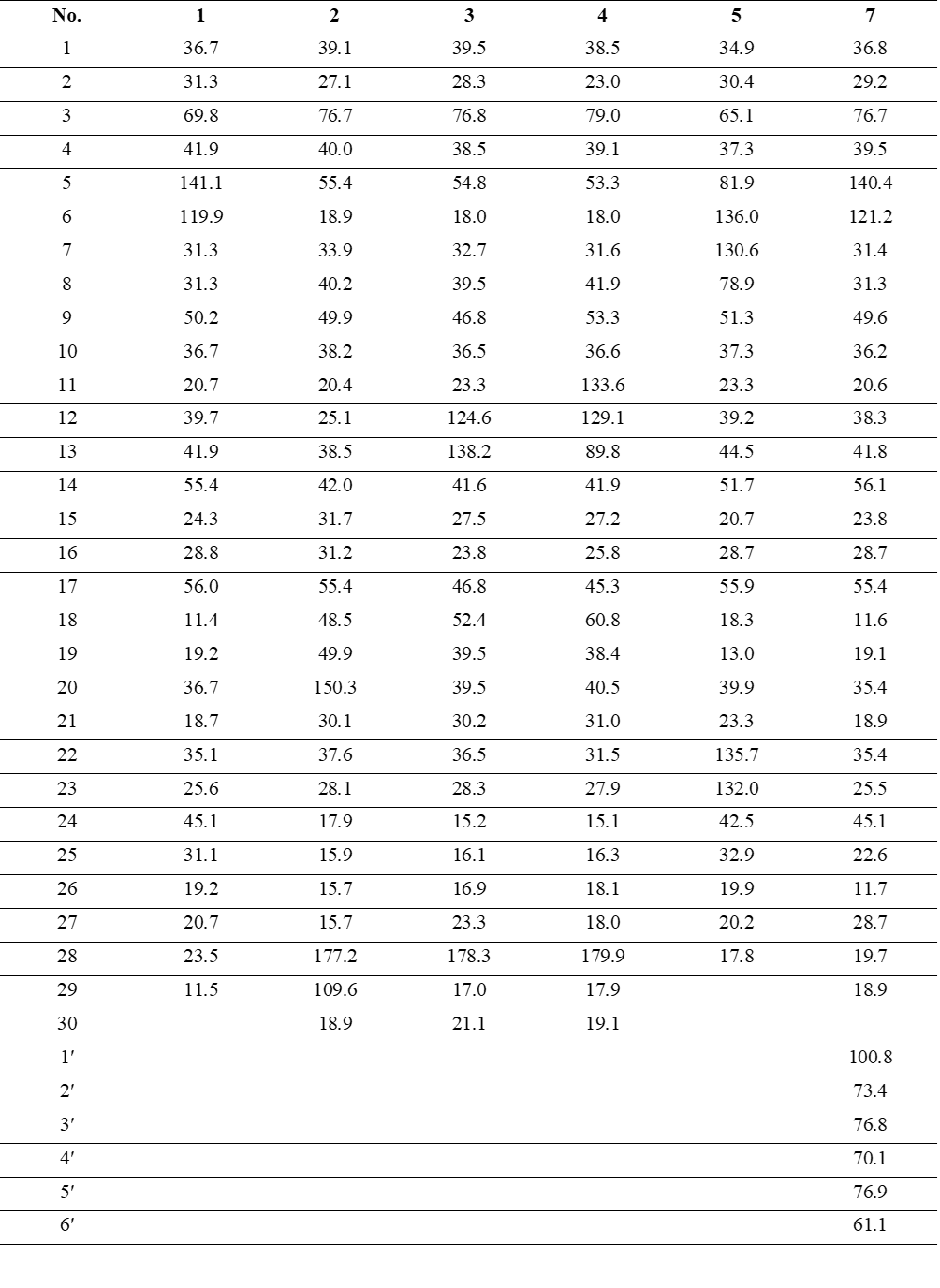
Table 3 1H (600 MHz) and 13C (150 MHz) NMR data of compound 6 in DMSO-d6 (δ in ppm, J in Hz).
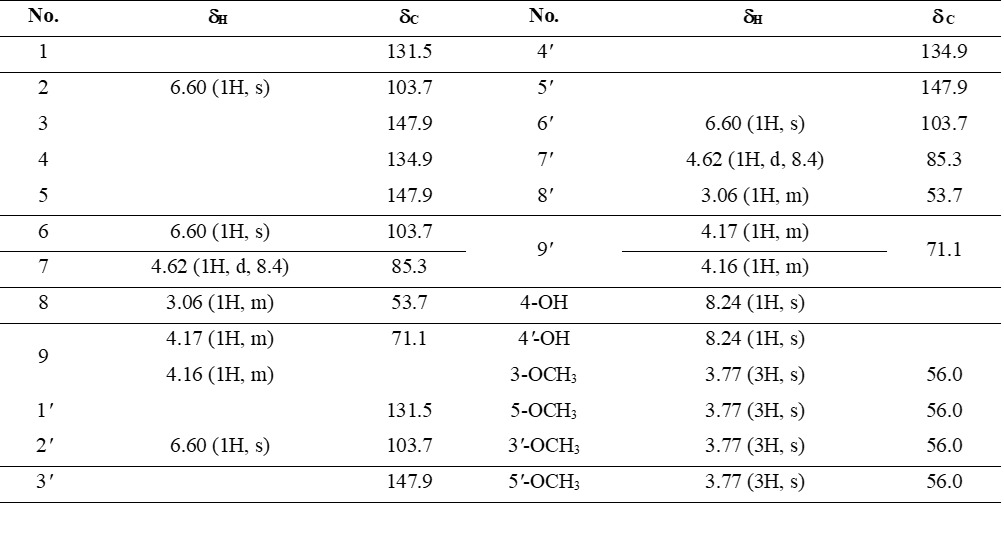
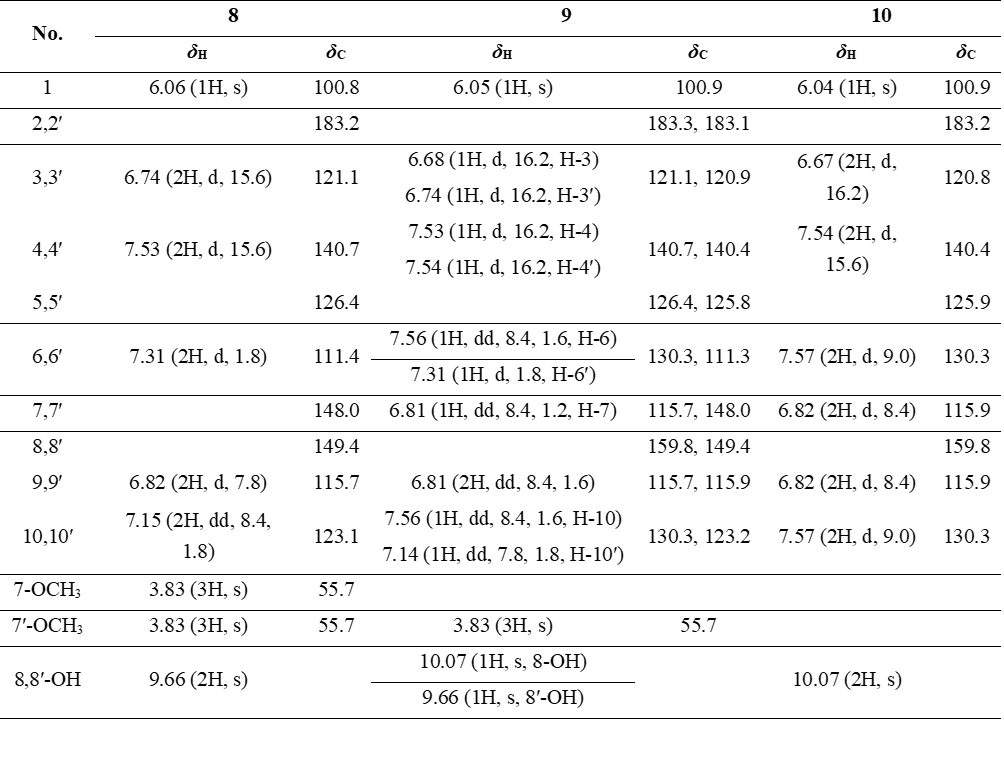
Table 4 1H (600 MHz) and 13C (150 MHz) NMR data of compounds 8 - 10 in DMSO-d6 (δ in ppm, J in Hz).
Bioactivities
The yeast α-glucosidase inhibitory activity of 1 - 10 was evaluated (Table 5). The IC50 values of 2 - 10 ranged from 75.5 to 163.8 µM, indicating significant inhibitory activity. Among the tested compounds, compound 9 exhibited the most potent inhibitory activity (IC50 75.5 ± 0.06 µM), outperforming acarbose (IC50 198.9 ± 0.02 µM), whereas compound 1 was inactive (IC50 > 200 µM). It should be noted that the inhibitory potency of compounds 2 - 10 may vary when tested against mammalian intestinal α-glucosidases.
Compounds 1 - 10 were also assessed for xanthine oxidase inhibitory activity (Table 5). Only compound 8 exhibited weak inhibition (IC50 95.0 ± 0.16 µM), while compounds 1 - 7 and 9 - 10 demonstrated no inhibitory activity (IC50 > 100 µM) compared to allopurinol (IC50 18.0 ± 0.07 µM).
Table 5 Inhibitory activities of compounds 1 - 10 against yeast α-glucosidase and xanthine oxidase.
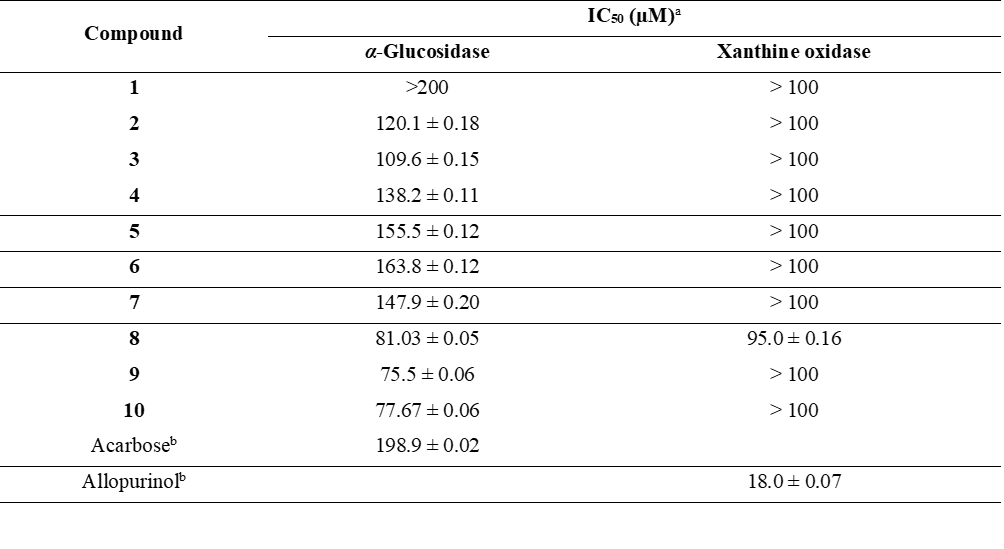
a IC50 values are presented as mean ± SD. b Positive control.
In silico prediction of binding modes, physicochemical, and pharmacokinetic properties of bioactive compounds
Given the strong α-glucosidase inhibitory activity of compounds 8 - 10 compared to the reference drug acarbose, molecular docking studies were performed to predict their binding interactions and affinities with the human enzyme. This approach is critical for evaluating their potential as antidiabetic agents acting locally in the intestine. The 3-dimensional structure of human intestinal α-glucosidase (PDB ID: 2QMJ) was used for docking to approximate clinically relevant conditions, even though the in vitro assays employed yeast α-glucosidase. Docking results revealed that the binding affinities of these compounds were consistent with their inhibitory trends. Compound 9 exhibited the lowest binding energy (–6.86 kcal/mol), suggesting slightly stronger interactions than compound 8 (–6.66 kcal/mol) and compound 10 (–6.74 kcal/mol). However, these differences (≤ 0.2 kcal/mol) fall within the typical computational uncertainty of docking methods and are not considered significant. Table 6 summarizes the binding affinities of compounds 8 - 10 compared with acarbose. Although acarbose showed a more favorable binding energy (–9.82 kcal/mol), indicating stronger theoretical interactions, the natural compounds still demonstrated meaningful binding within the enzyme’s active site, supporting their potential as alternative α-glucosidase inhibitors.
Table 6 Binding affinities of compounds 8 - 10 and acarbose against human intestinal α-glucosidase (PDB ID: 2QMJ).

It was found that the molecular docking poses of compounds 8 - 10, each featuring a diarylheptanoid scaffold, exhibit a high degree of similarity within the α-glucosidase active site, suggesting a conserved binding mode among these analogs (Figure 2).
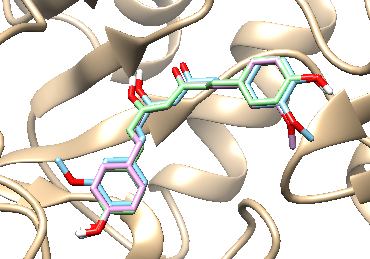
Figure 2 Binding modes of compounds 8 - 10 and acarbose within the α-glucosidase active site: superimposed docking poses of 8 (blue), 9 (pink), and 10 (green).
Two-dimensional interaction diagrams of compounds 8 - 10 (Figure 3) clearly illustrate both the interacting functional groups of the compounds and the corresponding amino acid residues. All 3 compounds utilize the central β-diketone moiety (in its enol or ketone form) to form hydrogen bonds with α-glucosidase. Additionally, hydroxyl groups from the aromatic rings contribute to hydrogen bonding, with compound 8 involving hydroxyl groups from 2 aromatic rings. Notably, compounds 8 - 10 exhibited highly similar interaction profiles. Arg526 and Gln603 were common hydrogen-bonding residues across all 3 compounds, while compound 8 additionally interacted with Lys480, and compounds 9 and 10 shared Asp542. Importantly, Arg526 was consistently involved in hydrogen bonding across all their compounds, indicating a conserved interaction at this key residue. In terms of hydrophobic interactions, each compound engaged multiple residues, with 8 residues being common among them: Asp203, Tyr299, Trp406, Met444, Asn449, Phe450, Phe575, and Tyr605. Among these, Asp203, Tyr299, Trp406, Met444, and Phe575 were also found to interact with acarbose (Table 7), indicating a conserved binding pattern between the active compounds and the reference inhibitor.
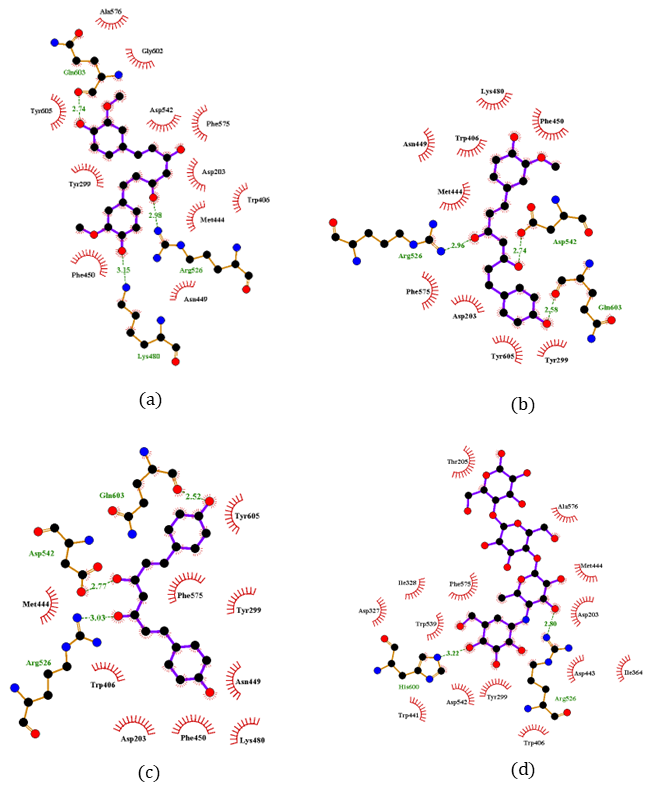
Figure 3 2D interaction diagrams of compounds 8 (a), 9 (b), 10 (c), and acarbose (d).
Table 7 Interaction profile of human intestinal α-glucosidase with compounds 8 - 10 and acarbose: Hydrophobic and hydrogen bonding residues.

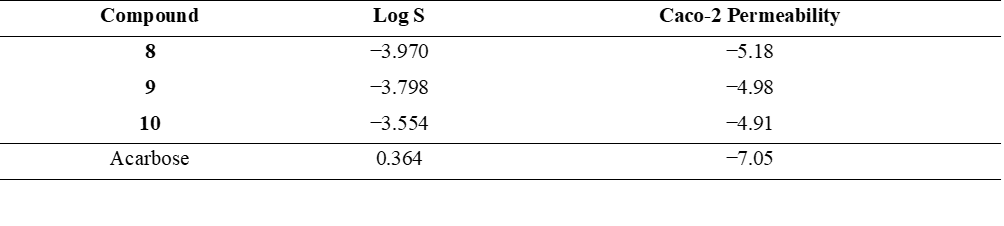
Although the natural compounds 8 - 10 demonstrated meaningful binding within the enzyme’s active site, further studies are required for the design of oral antidiabetic agents aimed at reducing carbohydrate absorption through α-glucosidase inhibition, similar to the mechanism of acarbose [24], it is essential to assess their solubility and intestinal permeability relative to acarbose. Upon oral administration, drug absorption starts with the disintegration of the dosage form, followed by the dissolution of the active pharmaceutical ingredient [25]. Once dissolved, the compound must reach the gastrointestinal tract where absorption occurs. Thus, both solubility and permeability are key determinants of oral bioavailability. These properties can be predicted using log S for aqueous solubility and Caco-2 permeability for intestinal absorption [16] (Table 8). An optimal log S range for orally absorbed drugs is typically between –4 and 0.5 log mol/L, [26] ensuring sufficient dissolution while maintaining membrane permeability. However, drugs that act locally in the gastrointestinal tract, such as α-glucosidase inhibitors, are designed not to be systemically absorbed. All 3 compounds (8 - 10) fall within or close to this range (–3.970 to –3.554), indicating adequate dissolution for local action, while acarbose shows much higher solubility (0.364). Regarding permeability, compounds with Caco-2 values greater than –5.15 log units are generally considered to have good intestinal absorption. Compounds 9 and 10 (–4.98 and –4.91) meet this criterion, whereas compound 8 (–5.18) is slightly below the threshold. In contrast, acarbose exhibits very low permeability (–7.05), consistent with its local mechanism. These findings suggest that compounds 9 and 10, with favorable solubility and moderate permeability, and compound 8, with slightly lower permeability, are promising candidates for development as locally acting oral antidiabetic agents.
Table 8 The logarithm of aqueous solubility and Caco-2 permeability of compounds 8 - 10 and acarbose.
Conclusions
From the fruits of Thai Helicteres isora L., 10 compounds were successfully isolated in this study, including 5 triterpenoids (1 - 5), 1 lignan (6), 1 triterpenoid glycoside (7), and 3 curcuminoids (8 - 10). Betulinic acid (2) is distributed in a variety of plant species [27]. Ursolic acid (3) has been widely isolated from various plant species [28], whereas 3β-hydroxyurs-11-ene-28(13)-lactone (4) has previously been reported from Hedyotis crassifolia L. [19]. Ergosterol-5,8-peroxide (5) has been isolated from Lobaria orientalis (Asahina) Yoshim. and Cladosporium anthropophilum, an endophytic fungus from Avicennia marina (Forssk.) Vierh. [29]. Syringaresinol (6) is primarily found in species of the genera Acanthopanax and Albizia [30], whereas β-sitosterol-3-O-β-D-glucoside (7) has been reported in Piper crocatum Ruiz & Pav., Paraboea leuserensis B.L. Burtt, and Crinum latifolium L. [22,31]. Curcumin (8), demethoxycurcumin (9), and bisdemethoxycurcumin (10) were isolated from turmeric (Curcuma longa L.) [23]. To the best of our knowledge, this is the first report of the isolation of compounds 2 - 10 from this species. Among all the isolated compounds, compound 9 exhibited the most potent α-glucosidase inhibitory activity, while compound 8 showed weak xanthine oxidase inhibition. Molecular docking revealed that compounds 8 - 10 have weaker binding affinities (–6.66 to –6.86 kcal/mol) compared with acarbose (–9.82 kcal/mol), yet they maintain meaningful interactions with key active-site residues such as Arg526, Trp406, and Phe575. Predicted solubility and intestinal permeability further support their potential as orally active, locally acting antidiabetic agents. These results highlight H. isora L. as a promising source of α-glucosidase inhibitory metabolites, warranting further investigation for therapeutic applications, particularly in diabetes management.
Acknowledgement
The authors gratefully acknowledge the financial support provided by Faculty of Science and Technology, Thammasat University, Thailand, Contract No. SciGR 8/2568.
Declaration of Generative AI in Scientific Writing
This manuscript employed generative AI tools, namely ChatGPT (OpenAI), exclusively to enhance language clarity, grammar, and readability under strict human supervision, without their use in generating scientific content, interpreting data, developing research questions, or drawing conclusions.
CRediT Author Statement
Phat Tan Mai: Investigation and Methodology. Lien Thi My Do: Investigation and Writing – Original Draft Preparation. Thuy Thi Le Nguyen: Methodology. Rico Ramadhan: Formal Analysis. Suwimon Khwunsiriwong: Methodology and Software. Thapakorn Chumphon: Formal Analysis. Jirapast Sichaem: Conceptualization, Resources, Data Curation, Project Administration, Funding Acquisition, and Writing – Review & Editing.
References
[1] TA Siswoyo, A Supriyadi, A Isnainun, E Novianti and R Harmoko. Impact of maturity stage on free radical scavenging and antidiabetic activities of Melinjo (Gnetum gnemon L.) seed proteins. Tropical Journal of Natural Product Research 2024; 8(8), 8001-8006.
[2] SMA Shah, F Akram, H Naeem, R Fatima, M Murtza, MS Aslam, AU Rehman and M Aamir. Investigation and evaluation of organic chemistry of alpha-glucosidase inhibitors for managing blood sugar levels. Indus Journal of Bioscience Research 2025; 3(6), 345-353.
[3] SK Liu, H Hao, Y Bian, YX Ge, S Lu, HX Xie, KM Wang, H Tao, C Yuan, J Zhang, J Zhang, CS Jiang and K Zhu. Discovery of new α-glucosidase inhibitors: Structure-based virtual screening and biological evaluation. Frontiers in Chemistry 2021; 9, 639279.
[4] N Kumar and AK Singh. Plant profile, phytochemistry and pharmacology of Avartani (Helicteres isora Linn.): A review. Asian Pacific Journal of Tropical Biomedicine 2014; 4(S1), S22-S26.
[5] KS Saravanan, R Gowri, KK Ramesh, S Shetty and S Maity. Potential use of Helicteres isora L. in diabetes mellitus: A systematic review of scientific literature. Pharmacognosy Communications 2023; 13(4), 163-168.
[6] K Beauty, S Talib and E Mohd. Phytochemistry and pharmacology of Helicteres isora Linn. (Marodphali): Ayurvedic insights and medicinal overview. International Journal of Ayurveda and Pharma Research 2024; 12(12), 118-128.
[7] S Pandey, D Patel, P Mishra and R Tiwari. Morphological, phytochemical and pharmacological study of Helicteres isora (Marorphali). International Journal of Research in Pharmacy and Pharmaceutical Sciences 2021; 6(3), 13-17.
[8] SP Mahire and SN Patel. Extraction of phytochemicals and study of its antimicrobial and antioxidant activity of Helicteres isora L. Clinical Phytoscience 2020; 6(1), 40.
[9] NH Nguyen, BP Vuong, CH Nguyen, AD Huynh, DM Nguyen, TH Duong and J Sichaem. Antimicrobial and α-glucosidase inhibitory compounds from the branches of Uvaria siamensis. Records of Natural Products 2024; 18(3), 331-338.
[10] TT Nguyen, TQ Tran, L Do, NM Huynh, K Inthanon and J Sichaem. α-Glucosidase and xanthine oxidase inhibitory activities from the fruits of Thai Averrhoa bilimbi L. Tropical Journal of Natural Product Research 2025; 9(3), 1105-1111.
[11] HM Berman, J Westbrook, Z Feng, G Gilliland, TN Bhat, H Weissig, IN Shindyalov and PE Bourne. The protein data bank. Nucleic Acids Research 2000; 28(1), 235-242.
[12] S Kim, J Chen, T Cheng, A Gindulyte, J He, Q Li, BA Shoemaker, PA Thiessen, B Yu, L Zaslavsky, J Zhang and EE Bolton. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Research 2021; 49, D1388-D1395.
[13] GM Morris, R Huey, W Lindstrom, MF Sanner, RK Belew, DS Goodsell and AJ Olson. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry 2009; 30(16), 2785-2791.
[14] EF Pettersen, TD Goddard, CC Huang, GS Couch, DM Greenblatt, EC Meng and TE Ferrin. UCSF Chimera - a visualization system for exploratory research and analysis. Journal of Computational Chemistry 2004; 25(13), 1605-1612.
[15] RA Laskowski and MB Swindells. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling 2011; 51(10), 2778-2786.
[16] L Fu, S Shi, J Yi, N Wang, Y He, Z Wu, L Zhang, X Liu, M Li and H Sun. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Research 2024; 52, W422-W431.
[17] RA El-Shiekh, DA Al-Mahdy, MS Hifnawy, T Tzanova, E Evain-Bana, S Philippot and EA Abdelsattar. Chemical and biological investigation of Ochrosia elliptica Labill. cultivated in Egypt. Records of Natural Products 2017; 11(6), 552-557.
[18] Noviany and H Osman. Structure elucidation of betulinic acid from Sesbania grandiflora root. Journal of Physics: Conference Series 2021; 1751, 012090.
[19] BC Huu and NKP Phung. Contribution to the study on chemical constituents of Hedyotis crassifolia L. (Rubiaceae). Vietnam Journal of Chemistry 2007; 45(3), 363-363.
[20] NTM Dung, PNKT Tuyen, J Mortier and NKPP Phung. Some triterpenoids and steroids from the lichen Lobaria orientalis, Lobariaceae. Journal of Science and Technology 2016; 54, 313-319.
[21] W Monthong, S Pitchuanchom, N Nuntasaen and W Pompimon. (+)-Syringaresinol lignan from new species Magnolia thailandica. American Journal of Applied Sciences 2011; 8(12), 1268-1271.
[22] NTT Nhung and VTB Huệ. Phân lập một số hợp chất tự nhiên từ thân hành Trinh nữ Hoàng cung (Crinum latifolium L., Amaryllidaceae) (in Vietnamese). Tạp chí Khoa học Lạc Hồng 2020; 9, 10-13.
[23] L Péret-Almeida, APF Cherubino, RJ Alves, L Dufossé and MBA Glória. Separation and determination of the physico-chemical characteristics of curcumin, demethoxycurcumin and bisdemethoxycurcumin. Food Research International 2005; 38(8-9), 1039-1044.
[24] FAV de Laar. Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vascular Health and Risk Management 2008; 4(6), 1189-1195.
[25] JB Dressman and J Krämer. Pharmaceutical dissolution testing. Taylor & Francis, New York, 2005.
[26] CA Lipinski, F Lombardo, BW Dominy and PJ Feeney. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development setting. Advanced Drug Delivery Reviews 2001; 46, 3-26.
[27] H Lu and Q Chen. A review on preparation of betulinic acid and its biological activities. Molecules 2021; 26, 5583.
[28] R Arulnangai, HA Thabassoom, HV Banu, K Thirugnanasambandham and R Ganesamoorthy. Recent developments on ursolic acid and its potential biological applications. Toxicology Reports 2025; 14, 101900.
[29] Y Mulyani, A Arofatus Naini, K Farabi, FF Abdullah, W Safriansyah, SE Sinaga, A Azhari, S Purbaya, A Peni Wulandari, S Fajriah, Y Shiono and U Supratman. The steroids produced by Cladosporium anthropophilum, an endophytic fungus isolated from Avicennia marina (Forssk.) Vierh and their antibacterial activity. Trends in Sciences 2024; 21(12), 8498.
[30] I Mssillou, M Bakour, FEZ Amrati, B Oraibi, ME Elmobark and A Khalid. Syringaresinol: Exploring its natural sources, bioactivity, and health benefits. Chemistry & Biodiversity 2025; 22, e00352.
[31] NL Harfita, A Santoni and S Suryati. Beta-sitosterol glycoside from Paraboea leuserensis and cytotoxicity test against MCF-7 human breast cancer cells. Riset Informasi Kesehatan 2023; 12(2), 272-276.
Supplementary Materials
Figure S1 Chemical structures of 1 - 10.
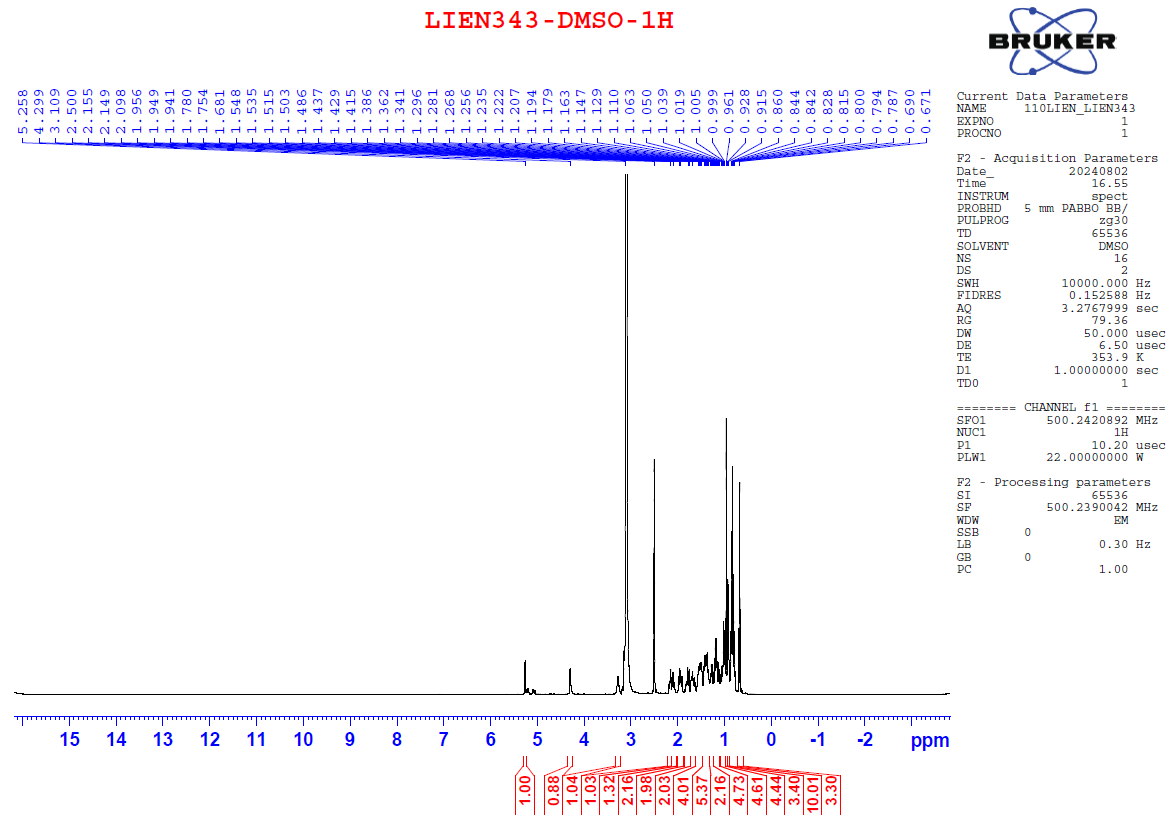
Figure S1.1 The 1H NMR spectrum of 1 in DMSO-d6.
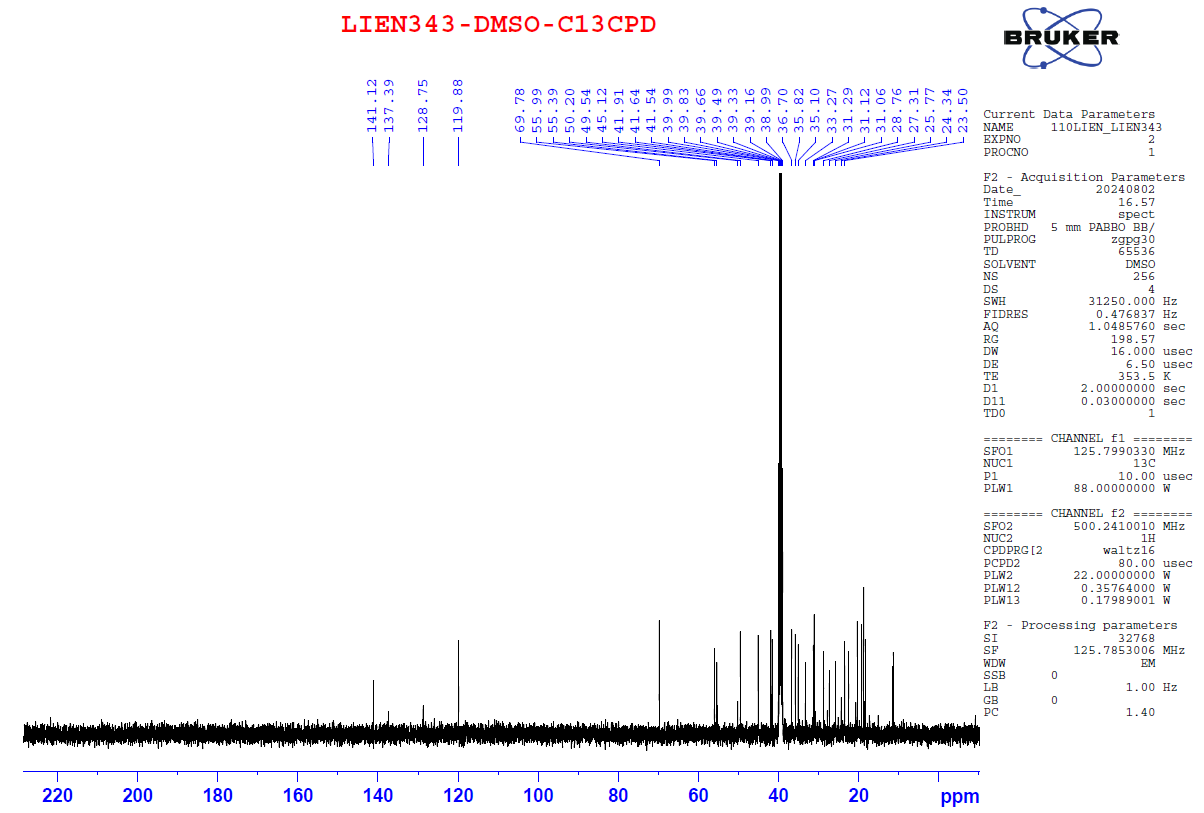
Figure S1.2 The 13C NMR spectrum of 1 in DMSO-d6.
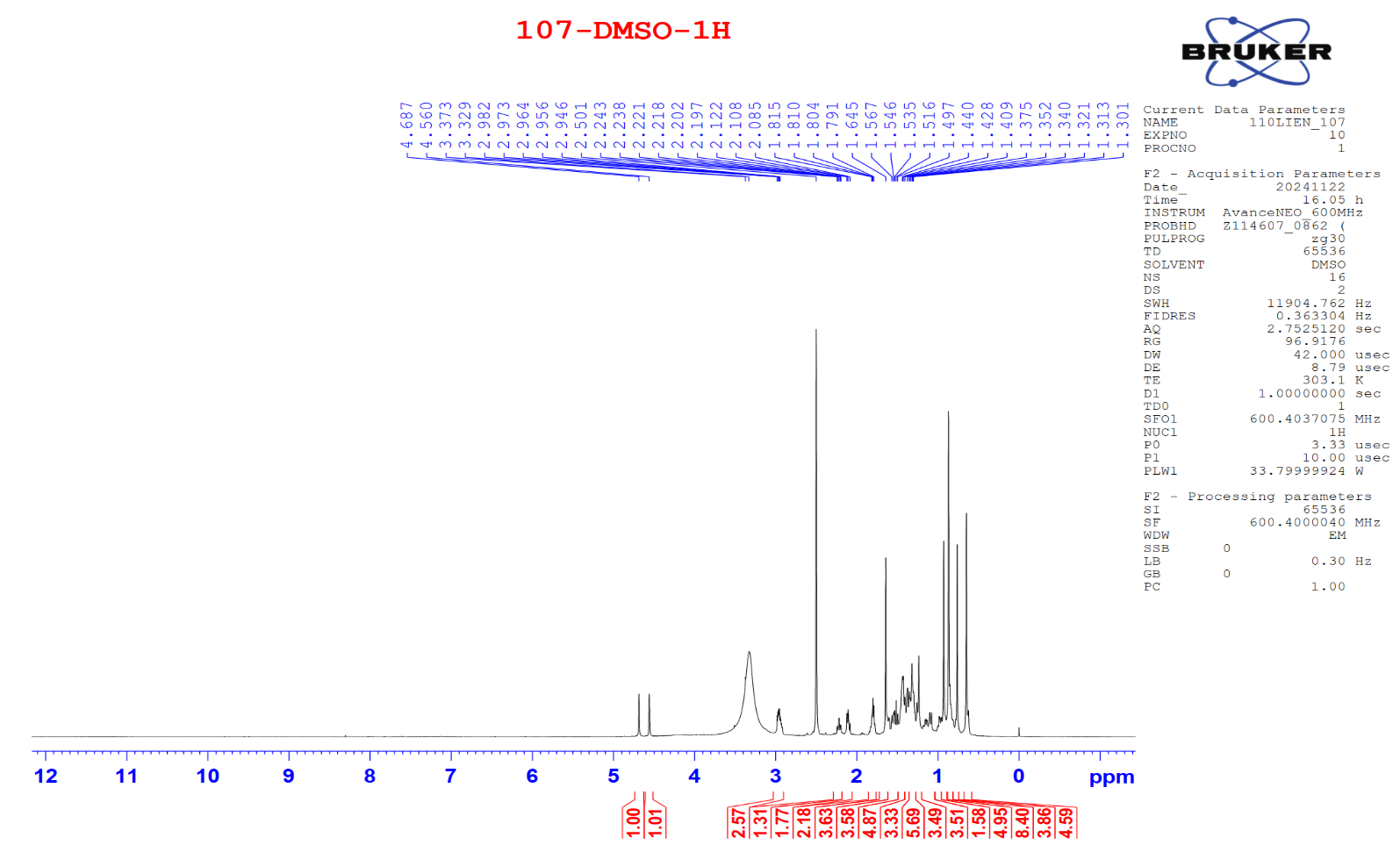
Figure S2.1 The 1H NMR spectrum of 2 in DMSO-d6.
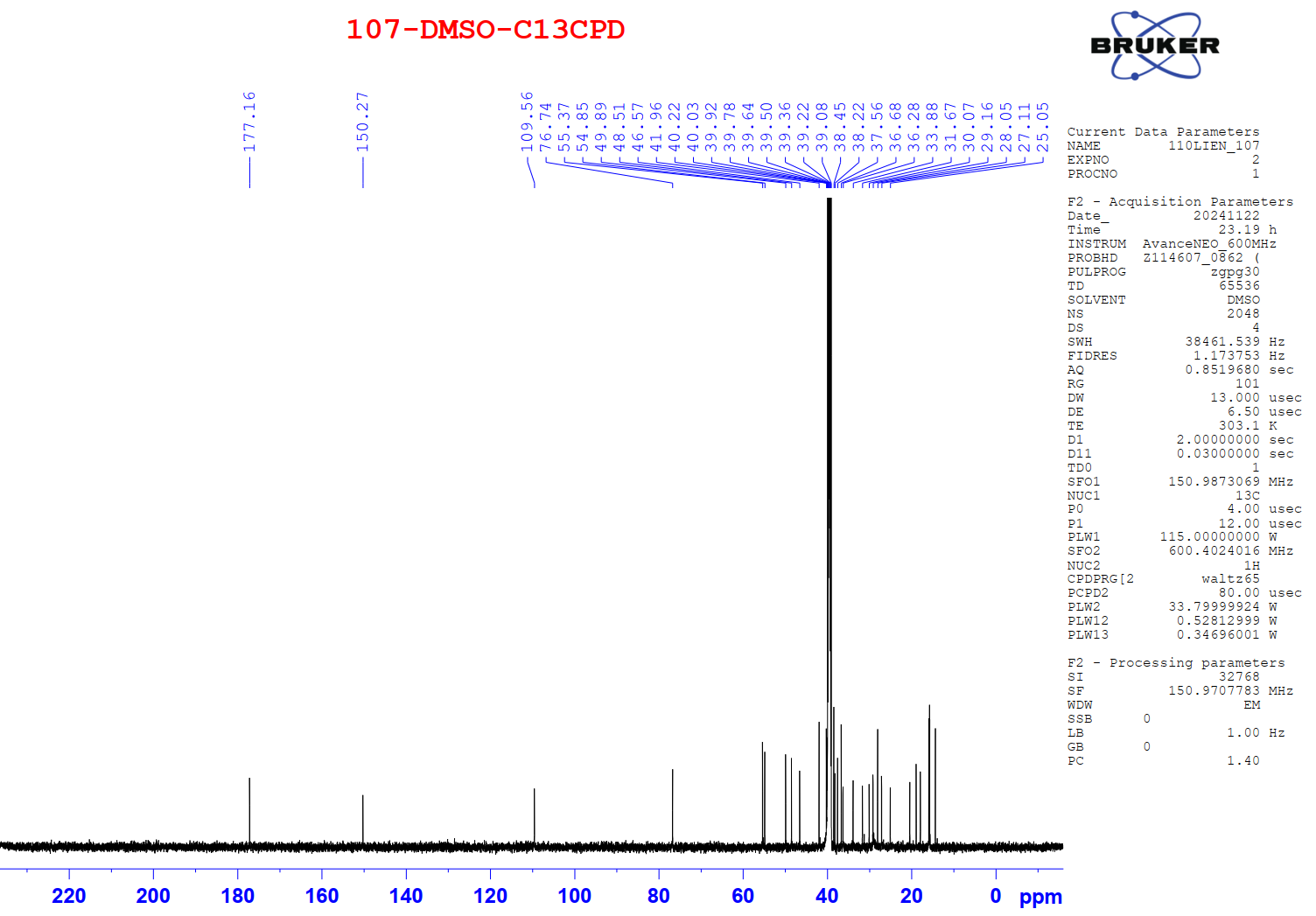
Figure S2.2 The 13C NMR spectrum of 2 in DMSO-d6.
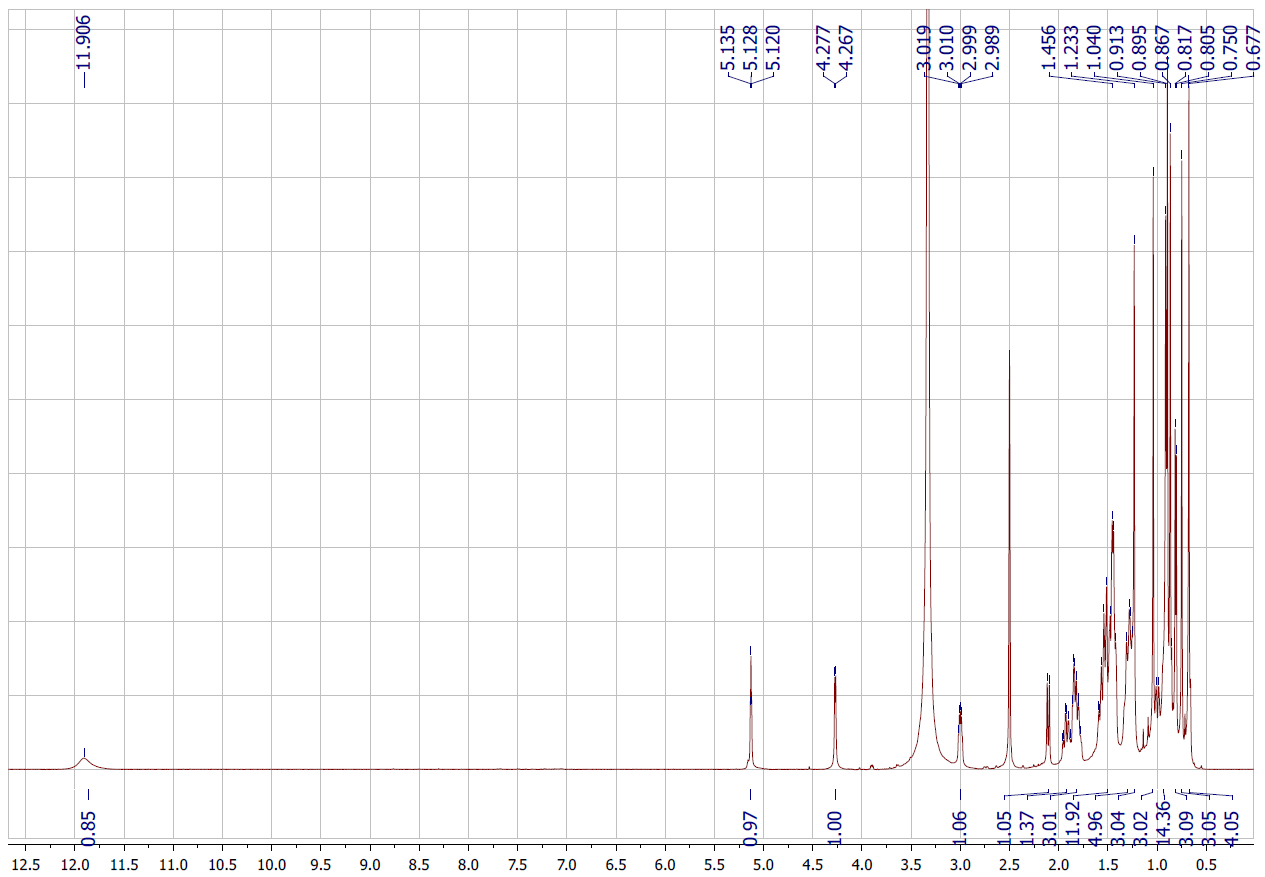
Figure S3.1 The 1H NMR spectrum of 3 in DMSO-d6.
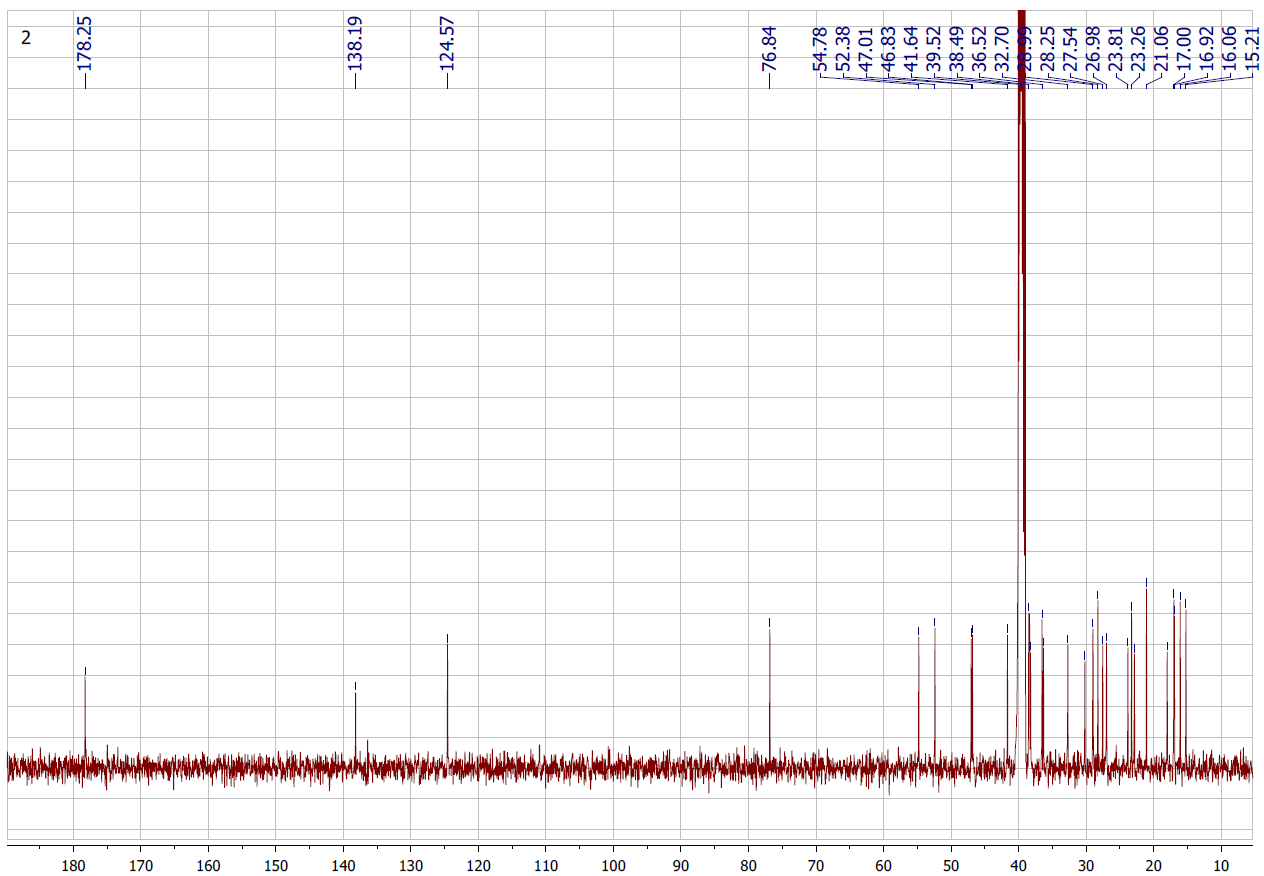
Figure S3.2 The 13C NMR spectrum of 3 in DMSO-d6.
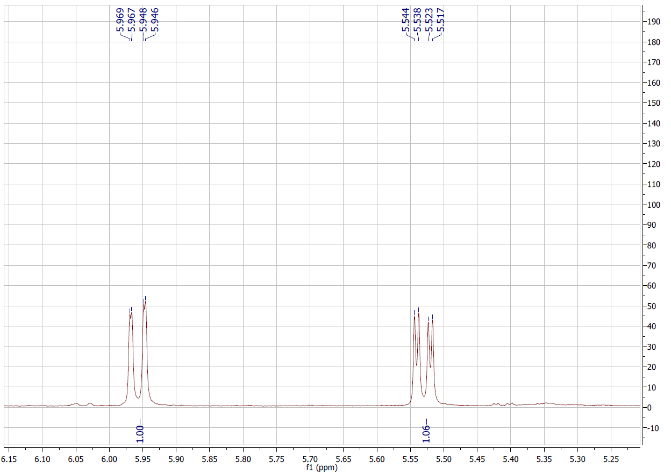
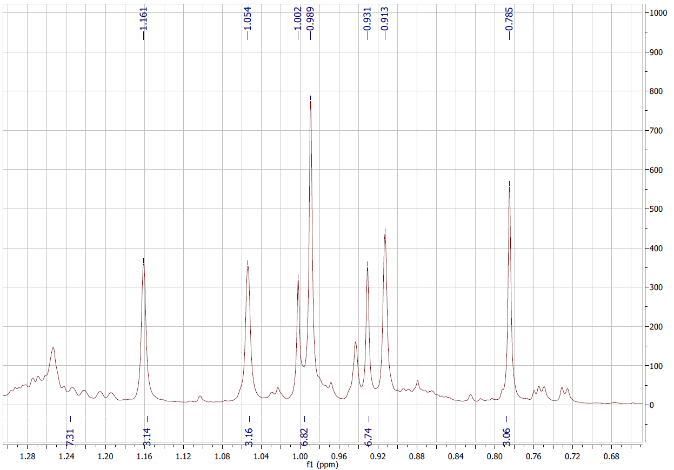
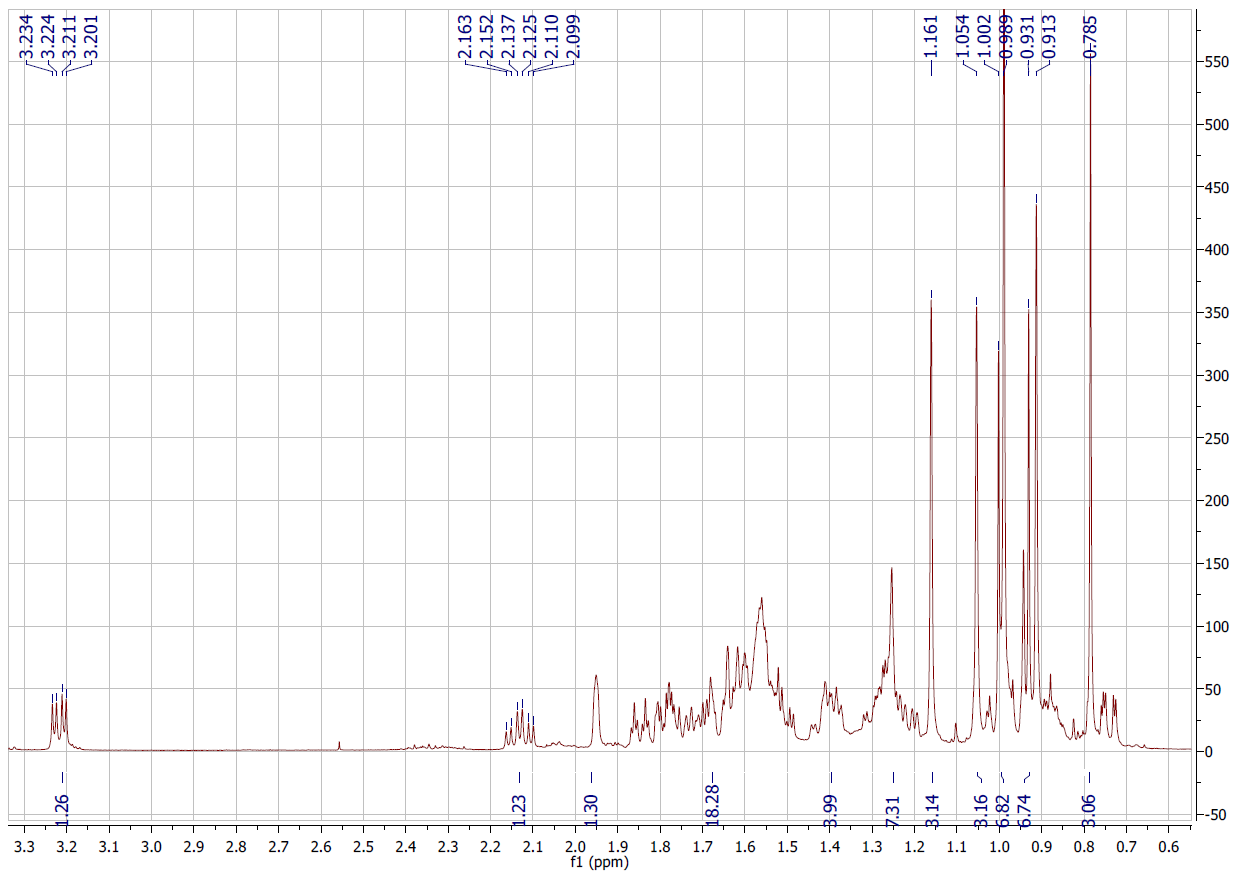
Figure S4.1 The 1H NMR spectrum of 4 in DMSO-d6.
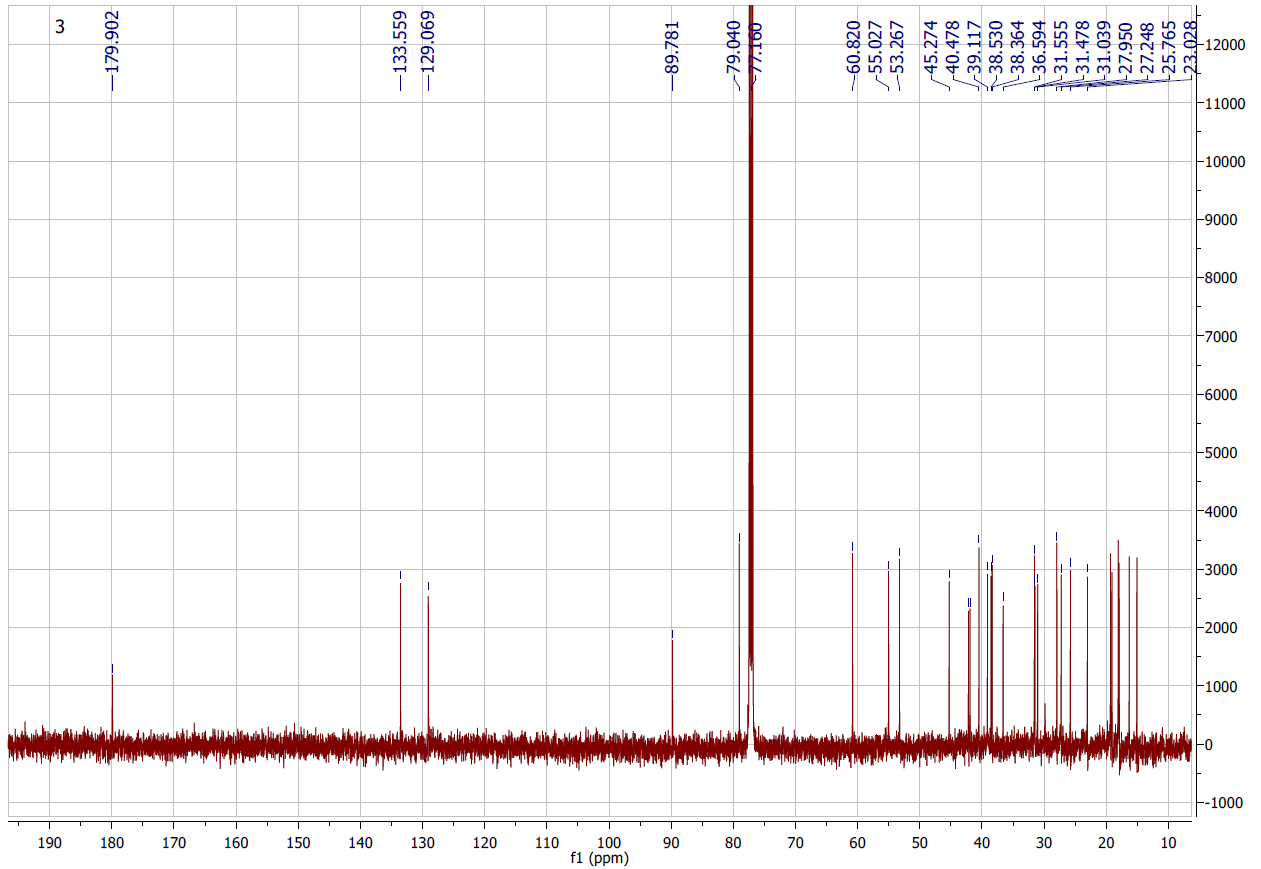
Figure S4.2 The 13C NMR spectrum of 4 in DMSO-d6.
Figure S4.3 The ESIMS spectrum of 4.
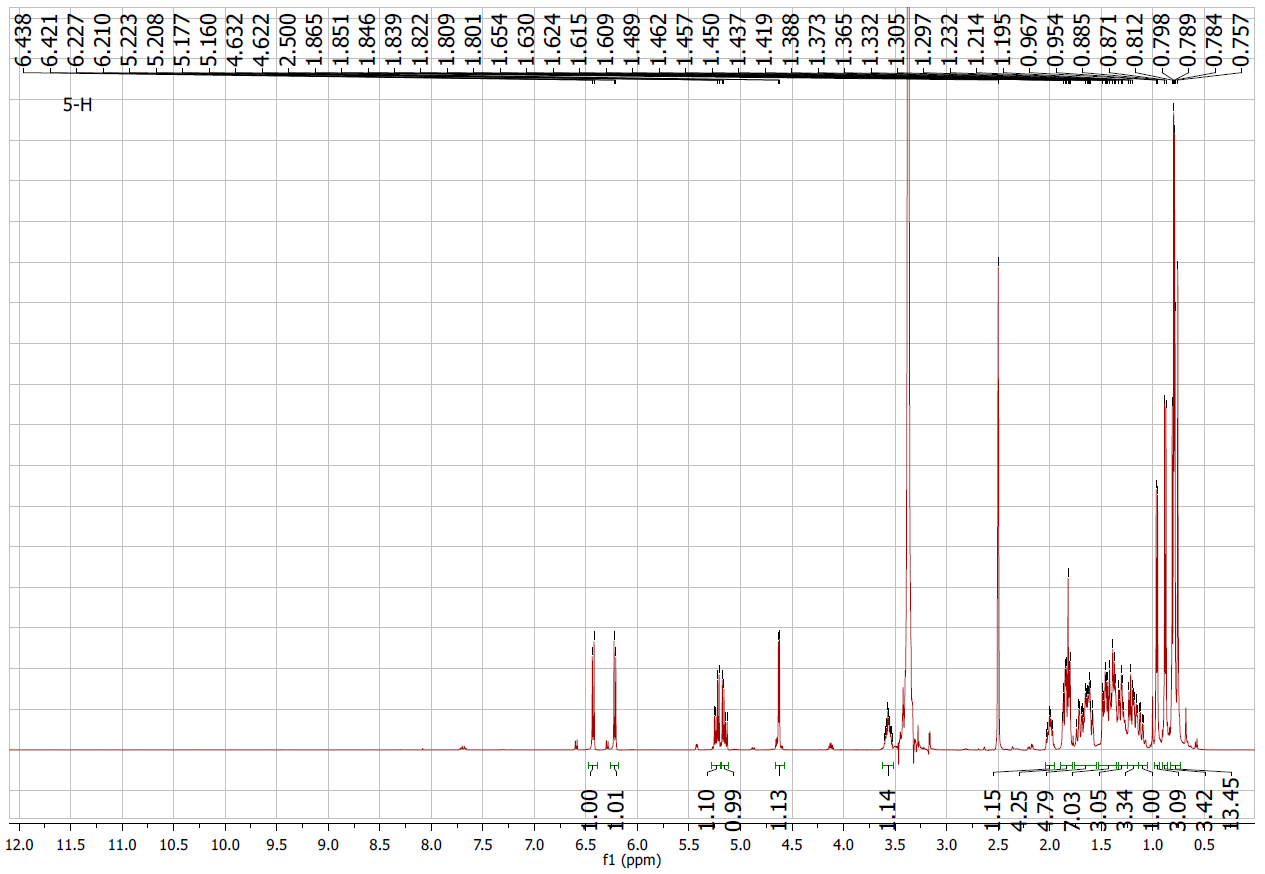
Figure S5.1 The 1H NMR spectrum of 5 in DMSO-d6.
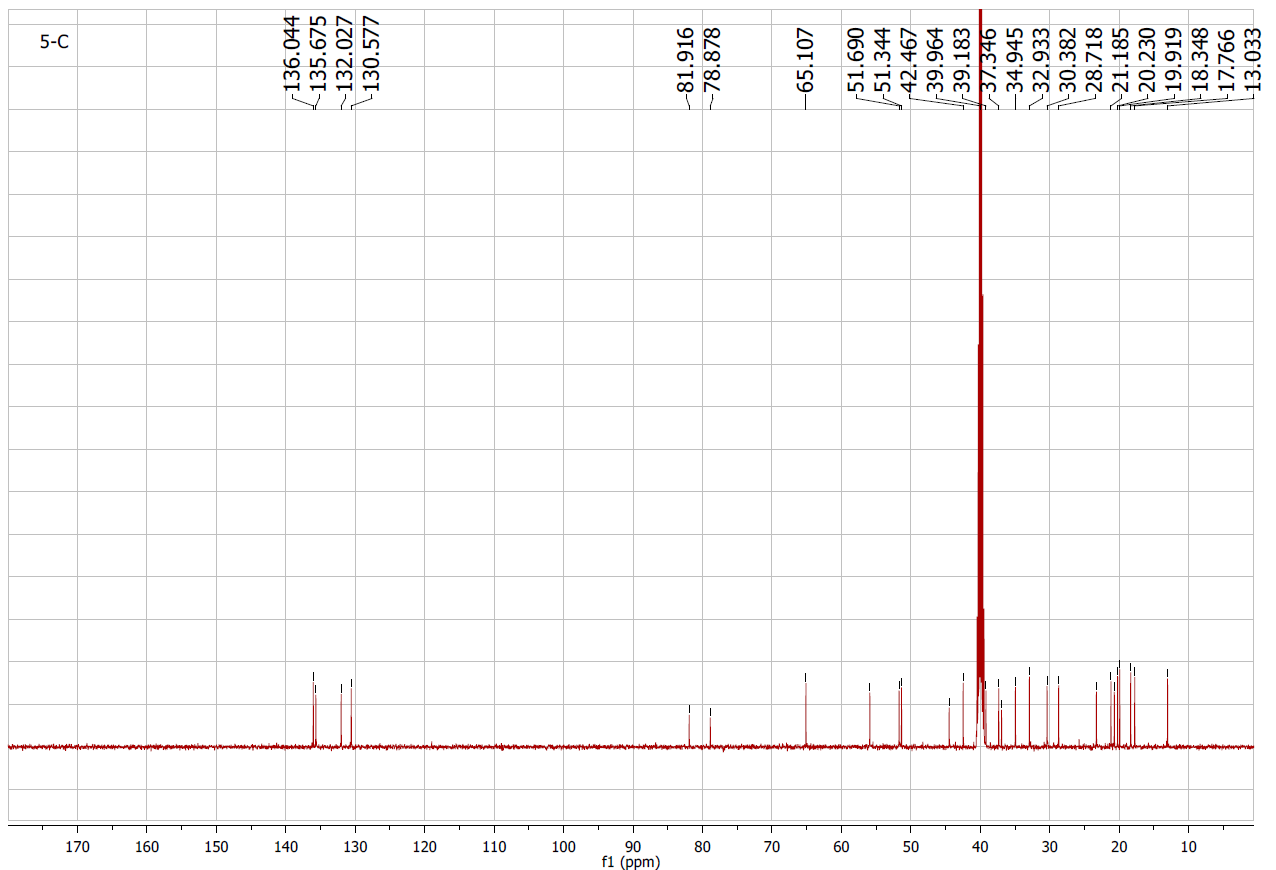
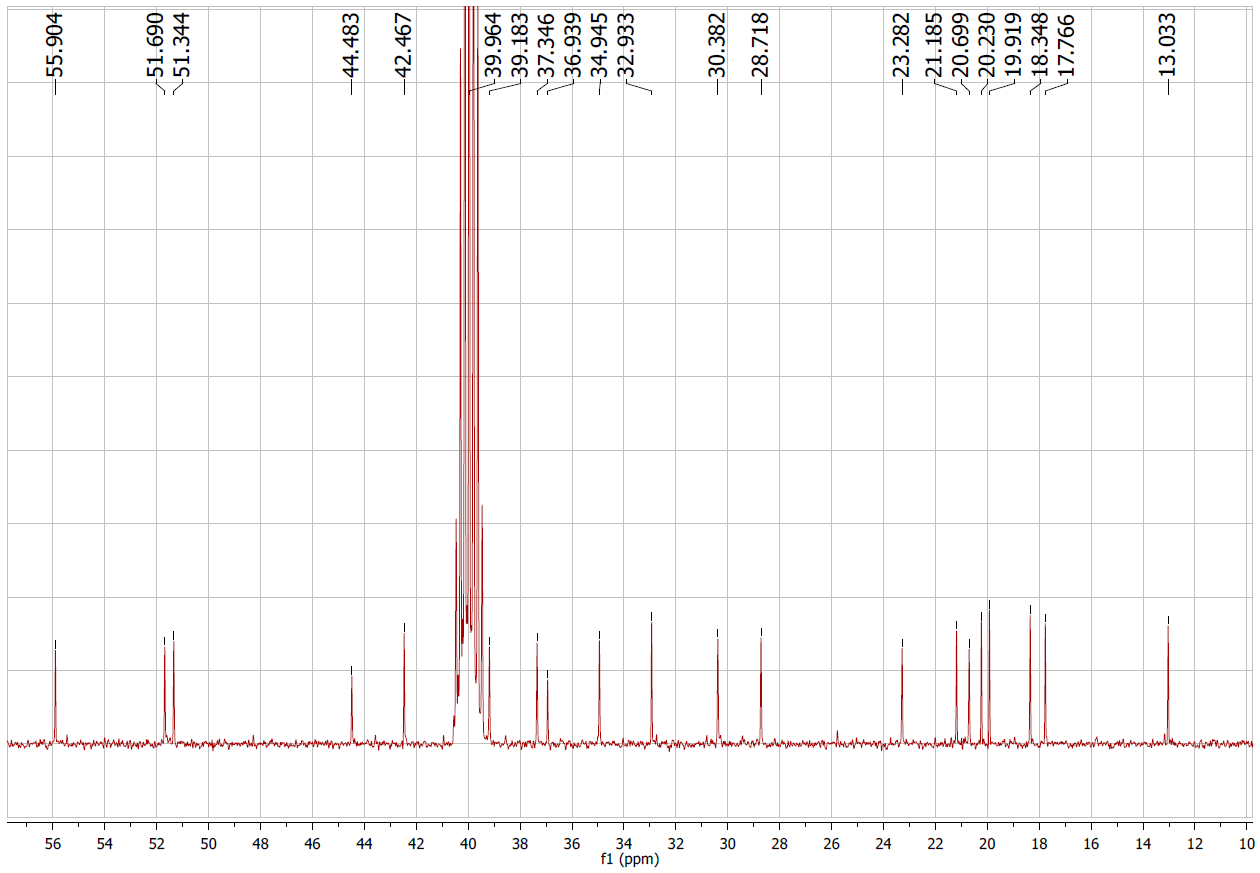
Figure S5.2 The 13C NMR spectrum of 5 in DMSO-d6.
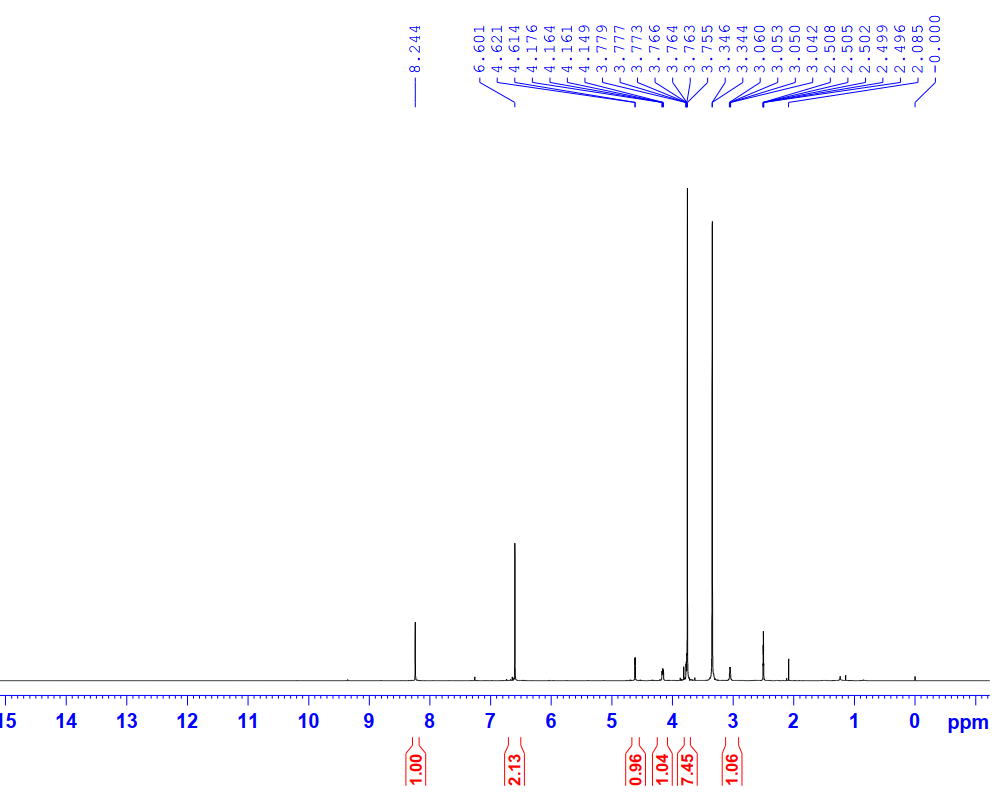
Figure S6.1 The 1H NMR spectrum of 6 in DMSO-d6.
Figure S6.2 The 13C NMR spectrum of 6 in DMSO-d6.
Figure S6.3 The HSQC spectrum of 6 in DMSO-d6.
Figure S6.4 The HMBC spectrum of 6 in DMSO-d6.
Figure S7.1 The 1H NMR spectrum of 7 in DMSO-d6.
Figure S7.2 The 13C NMR spectrum of 7 in DMSO-d6.
Figure S7.3 The HMBC spectrum of 7 in DMSO-d6.
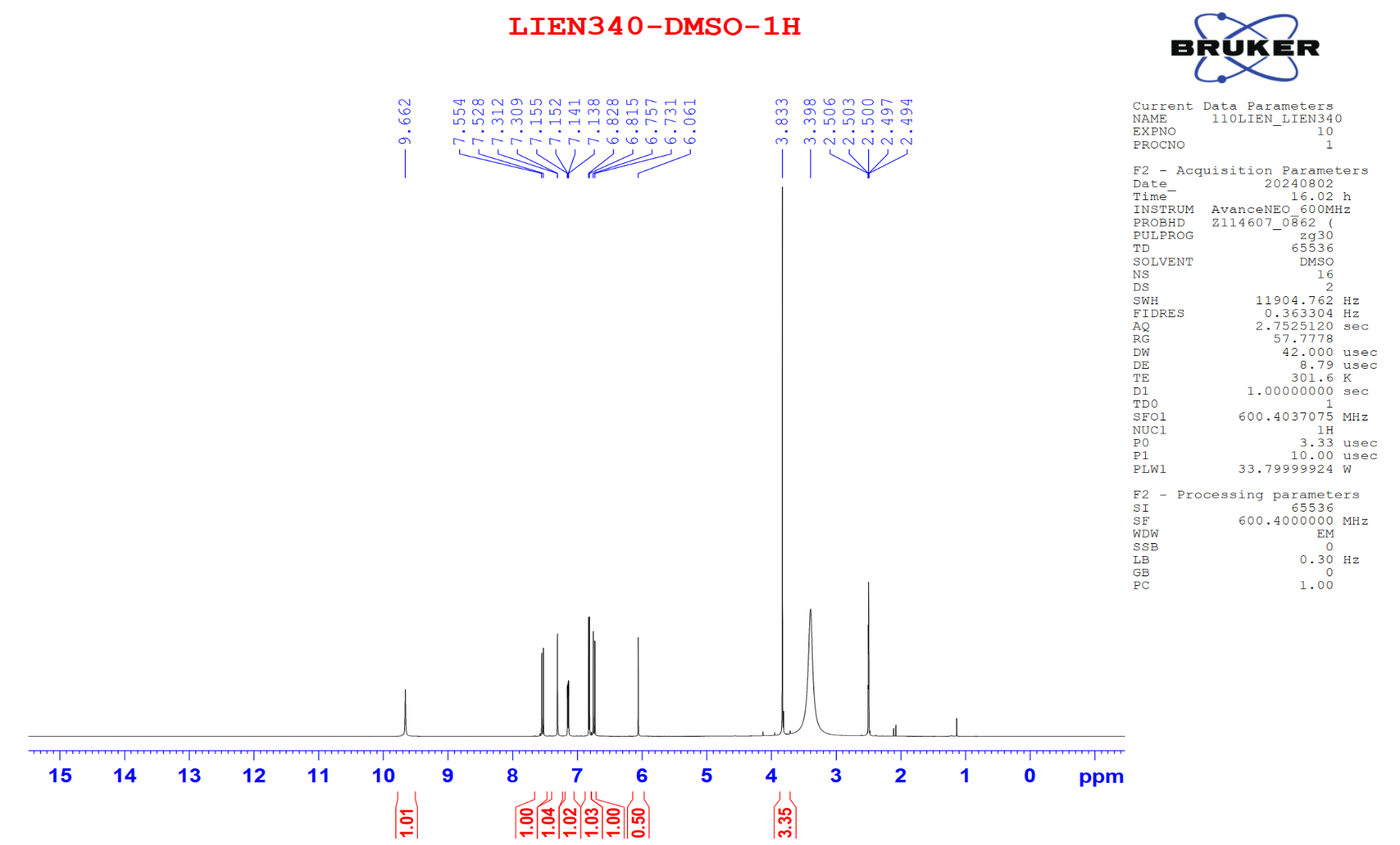
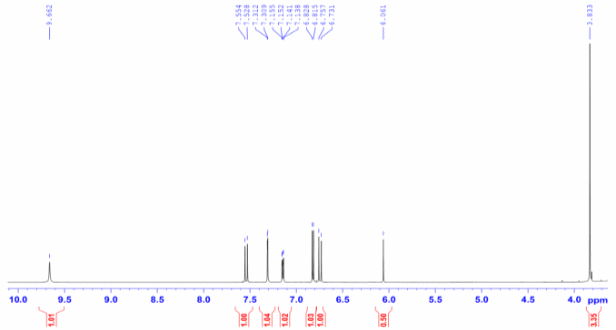
Figure S8.1 The 1H NMR spectrum of 8 in DMSO-d6.
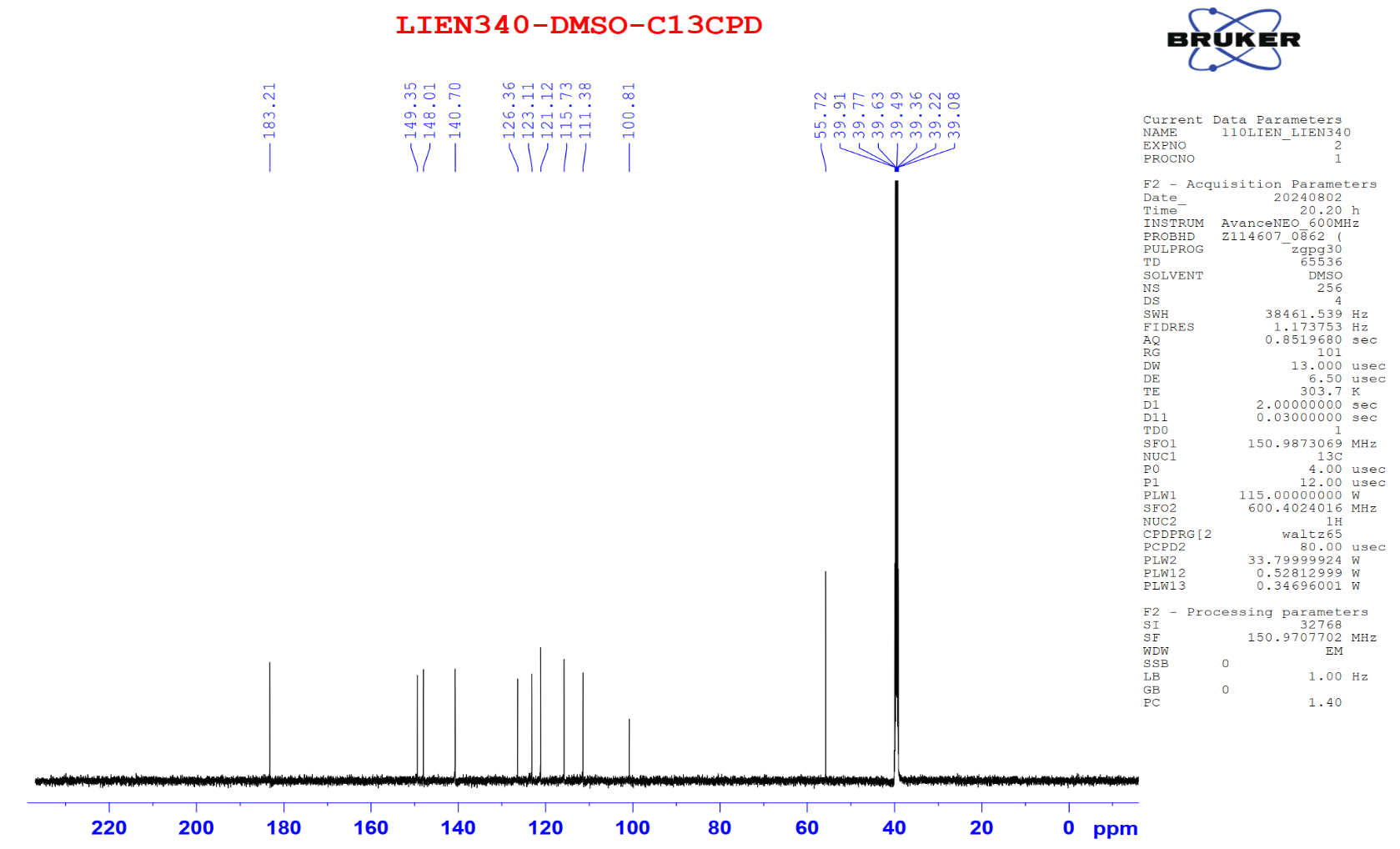
Figure S8.2 The 13C NMR spectrum of 8 in DMSO-d6.
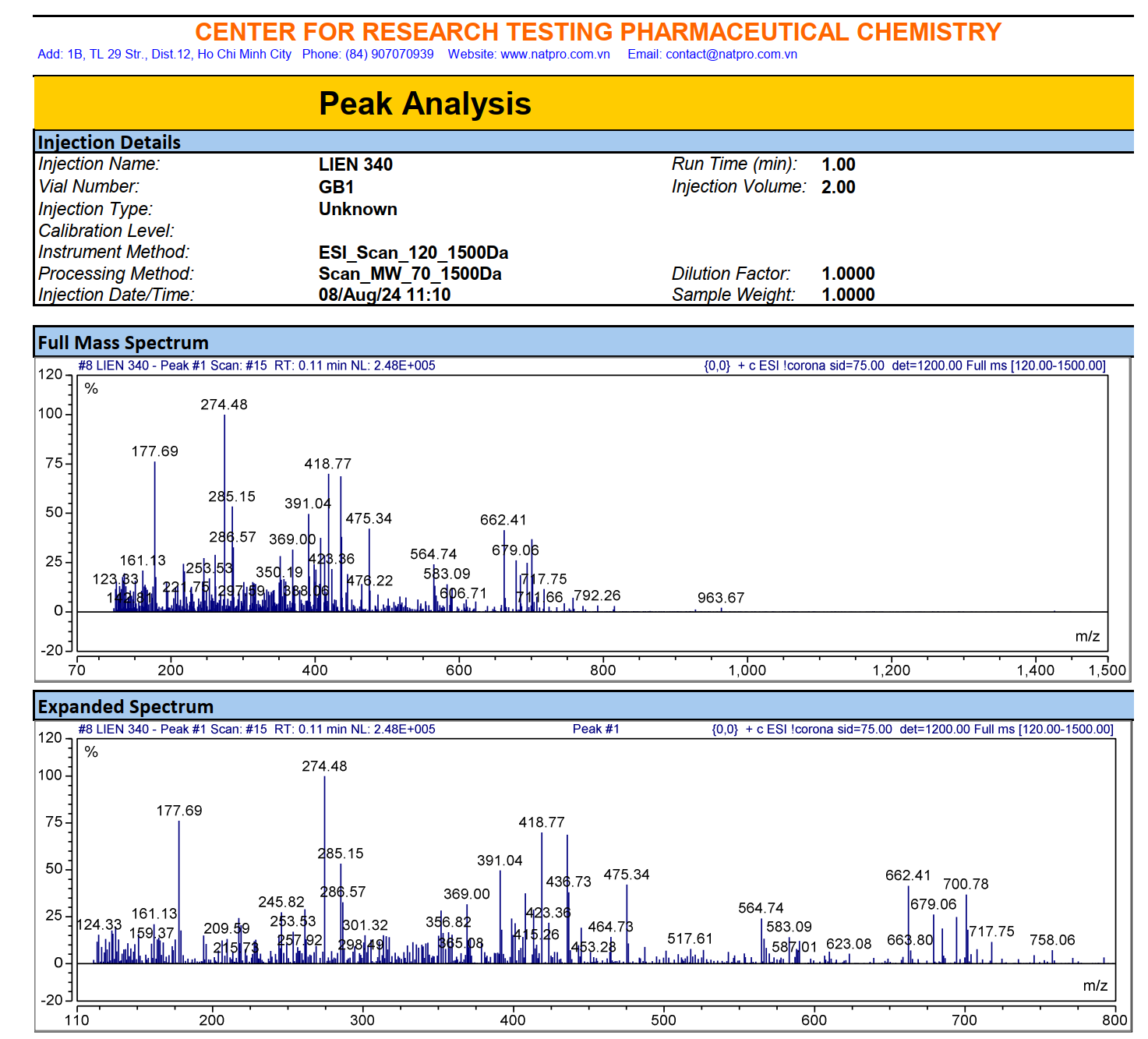
Figure S8.3 The ESIMS spectrum of 8.
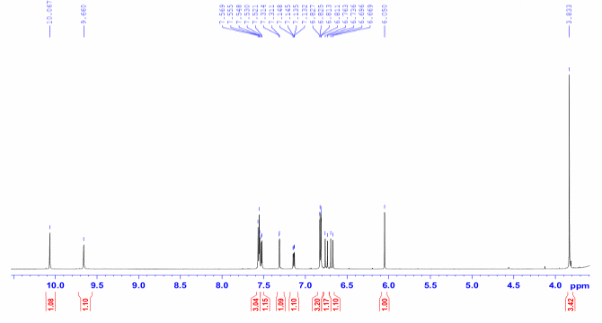
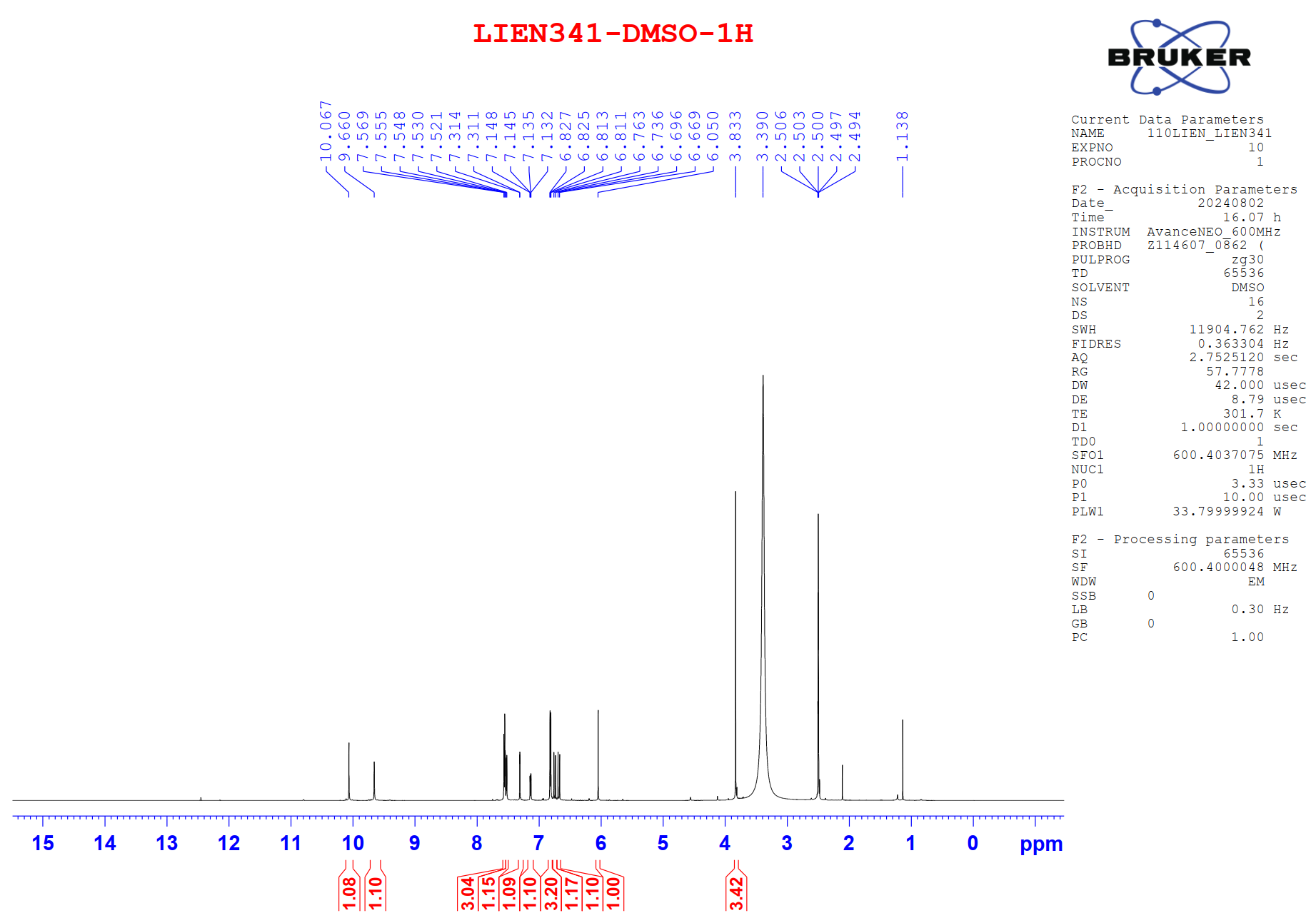
Figure S9.1 The 1H NMR spectrum of 9 in DMSO-d6.
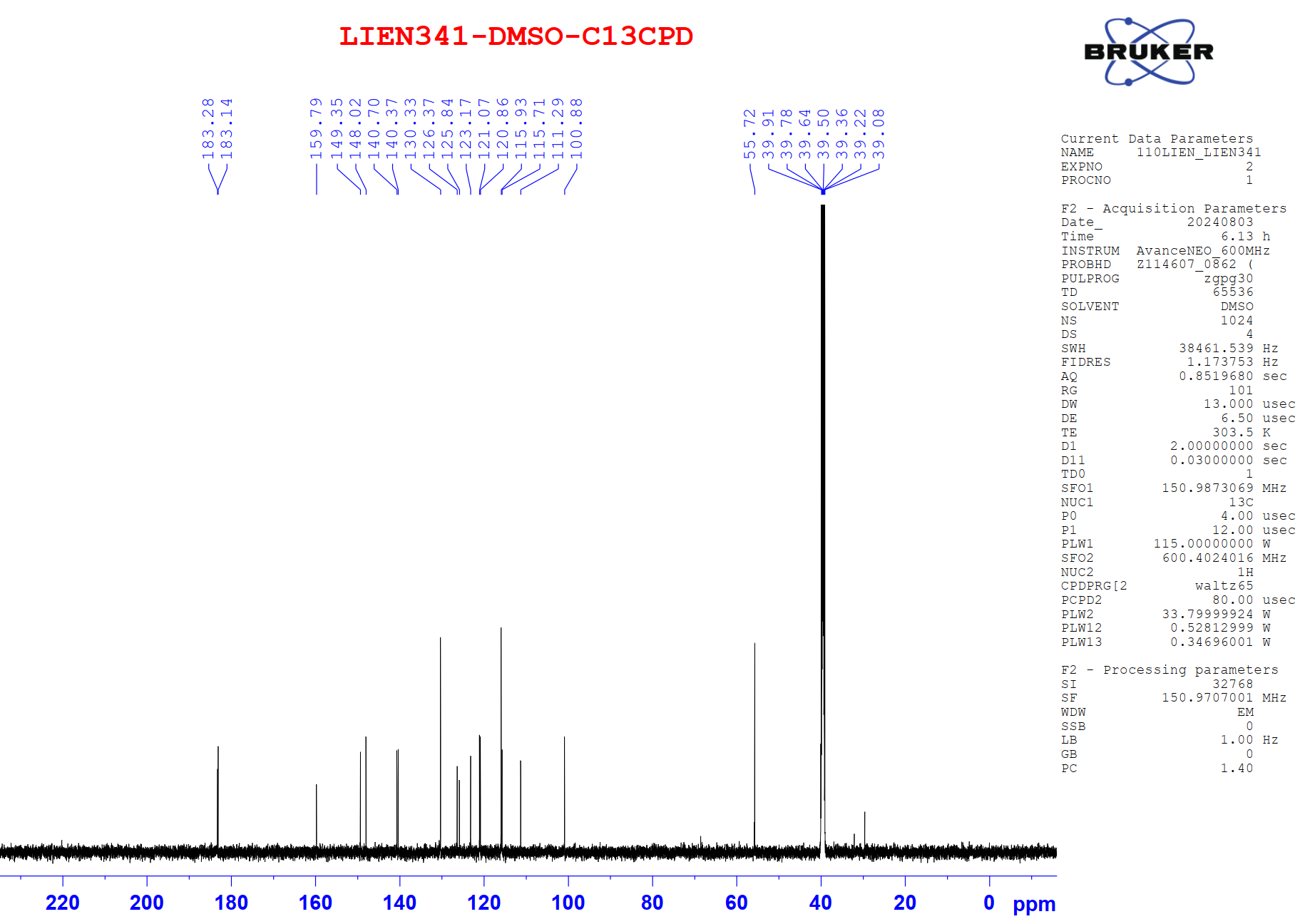
Figure S9.2 The 13C NMR spectrum of 9 in DMSO-d6.
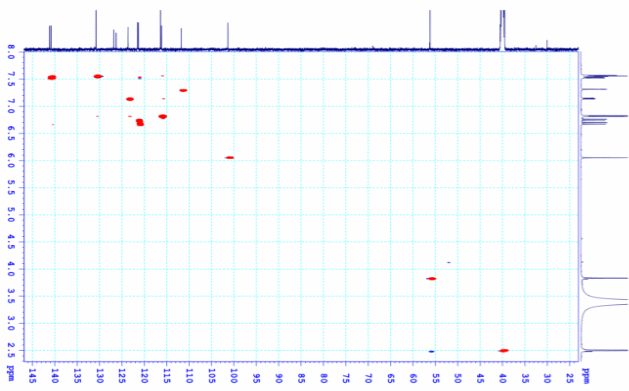
Figure S9.3 The HSQC spectrum of 9 in DMSO-d6.
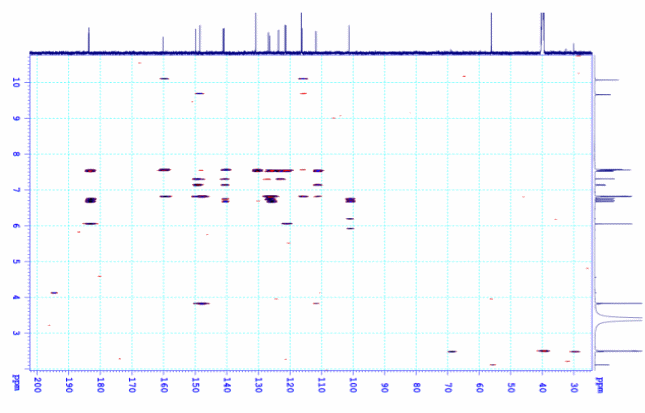 Figure
S9.4
The
HMBC spectrum of 9
in DMSO-d6.
Figure
S9.4
The
HMBC spectrum of 9
in DMSO-d6.
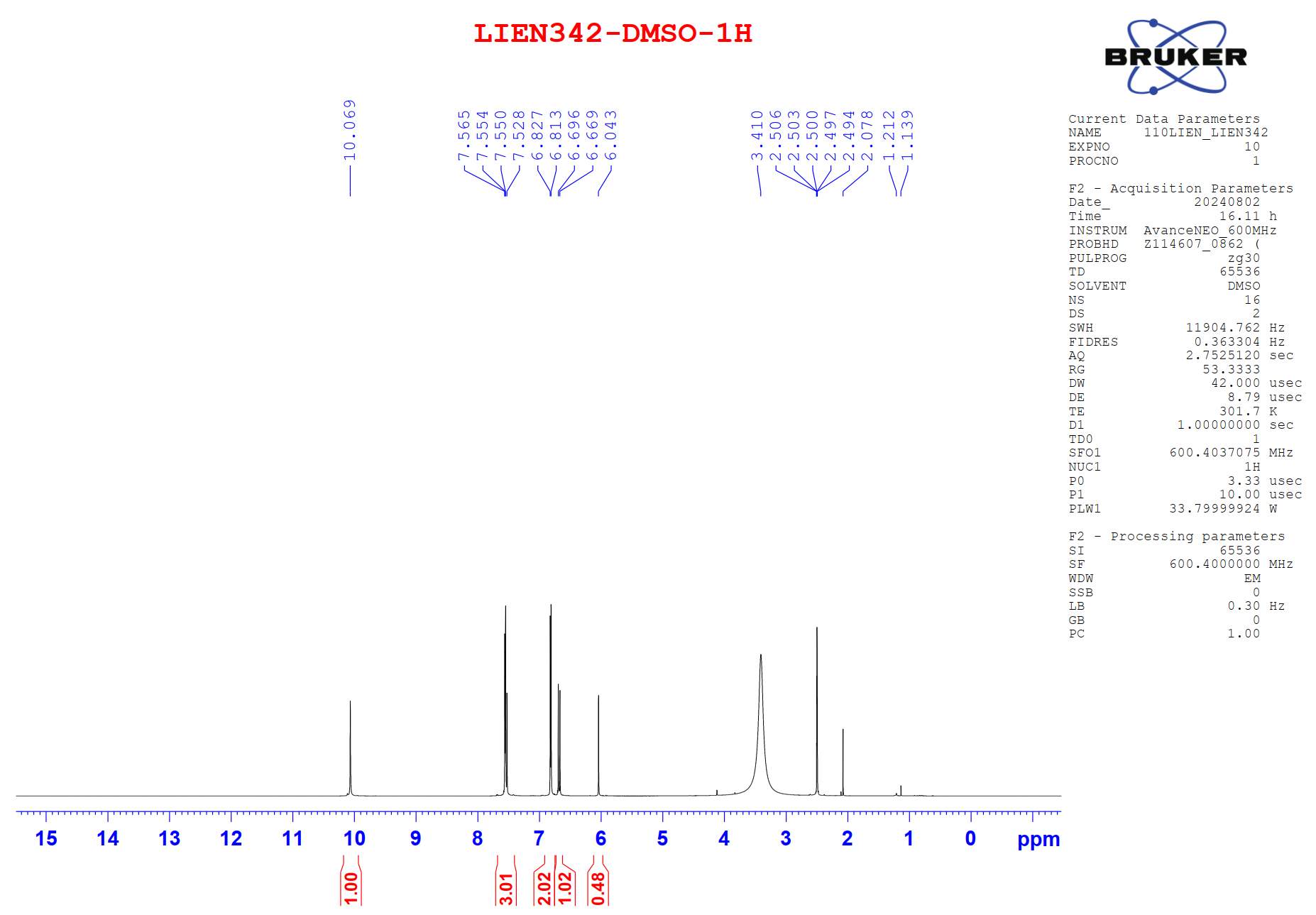
Figure S10.1 The 1H NMR spectrum of 10 in DMSO-d6.
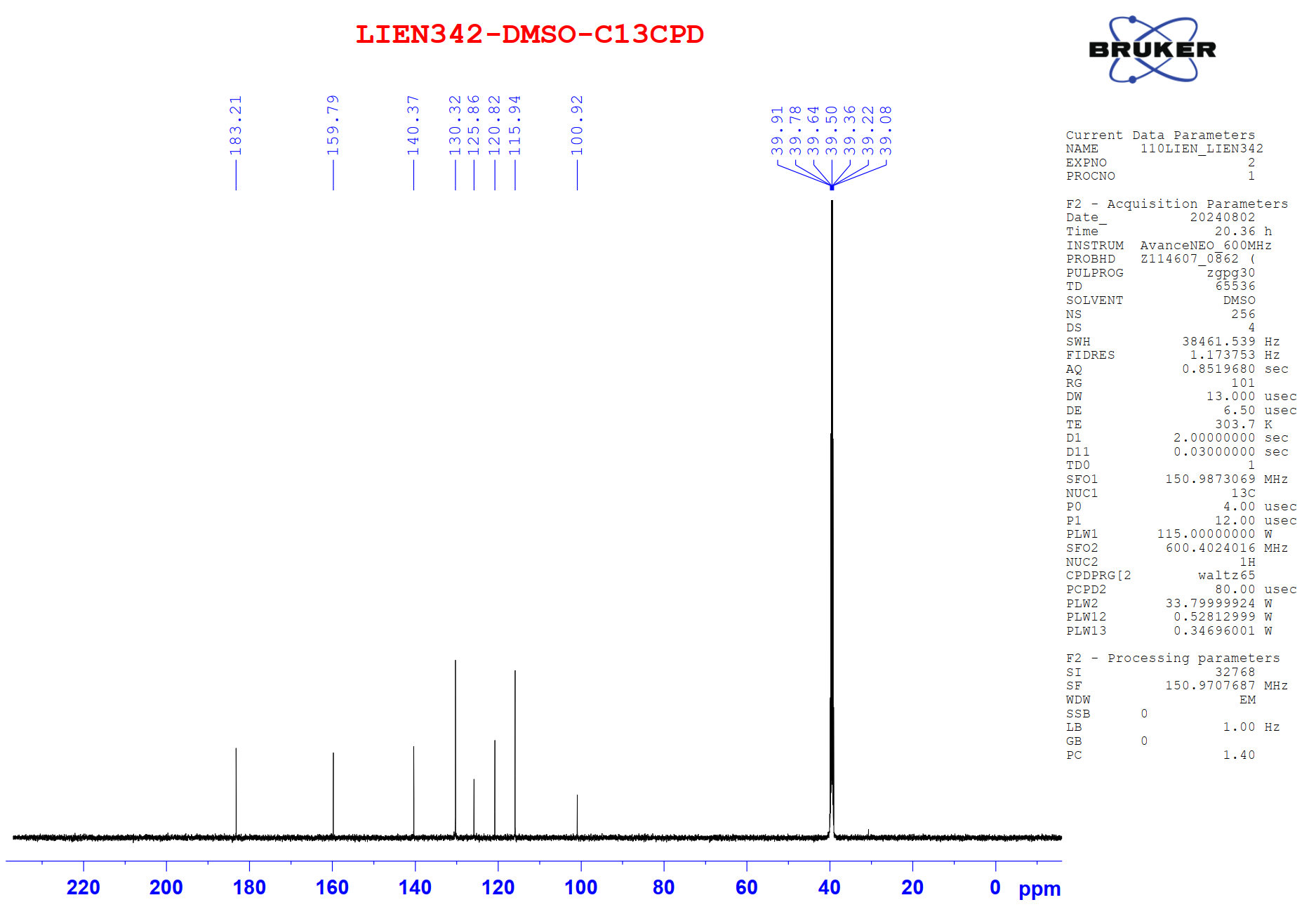
Figure S10.2 The 13C NMR spectrum of 10 in DMSO-d6.
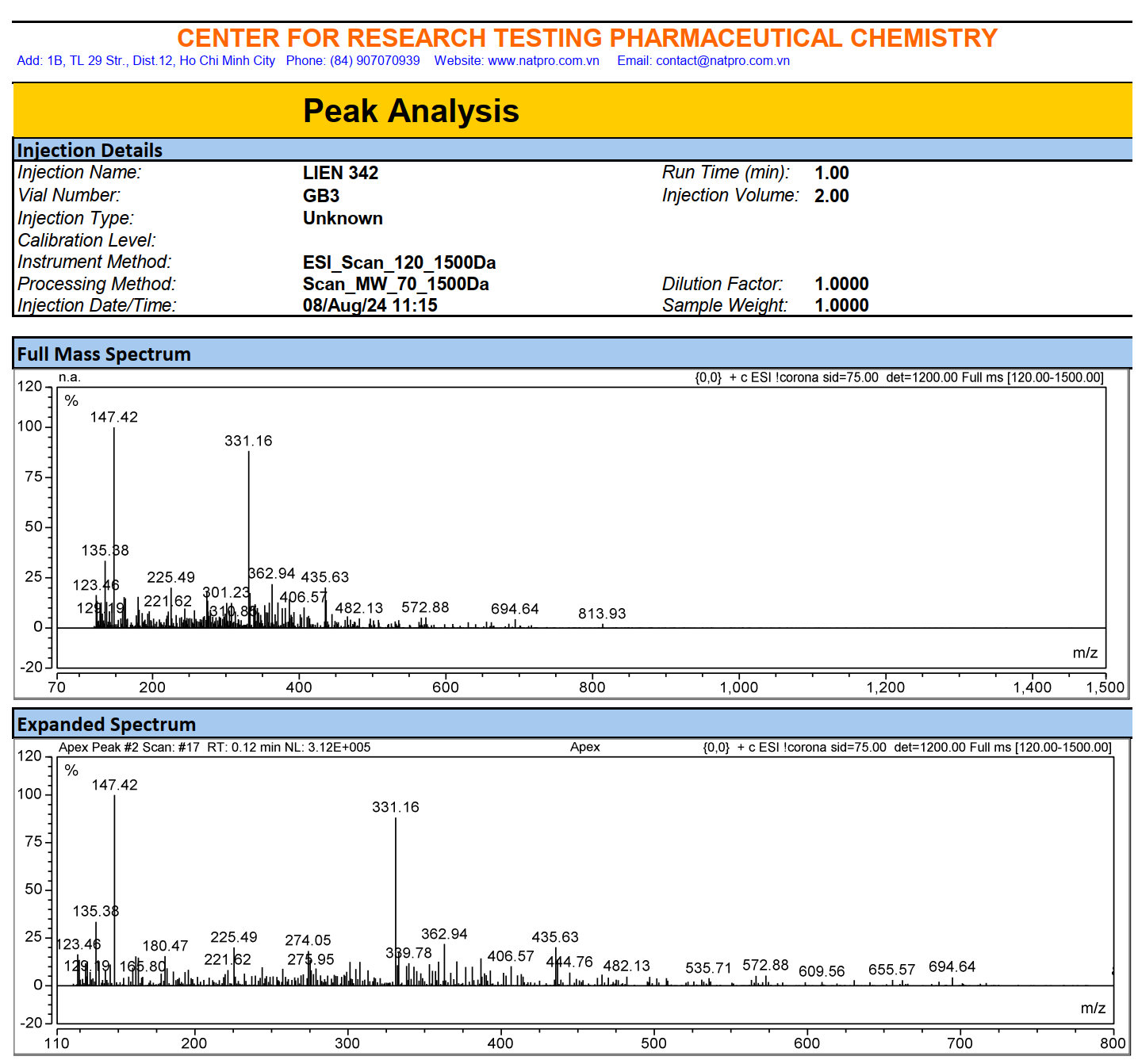
Figure S10.3 The ESIMS spectrum of 10.