Trends
Sci.
2026; 23(7): 12633
Molecular Docking and Pharmacokinetic Analysis of Antioxidant Content in Popular Indonesian Herbal Plants for Adjuvant Therapy in Cancer
Luqman Alwi1, Irwan Budiono1,*, Mahalul Azam1,
Selamat Budijitno2, Irena Intania1 and Matheus Prayoga Claus3
1Department of Public Health, Faculty of Medicine, Universitas Negeri Semarang, Jawa Tengah 50229, Indonesia
2Department of Oncology Surgery, Faculty of Medicine, Universitas Diponegoro, Jawa Tengah 50275, Indonesia
3Department of Pharmacy, Faculty of Medicine, Universitas Negeri Semarang, Jawa Tengah 50229, Indonesia
(*Corresponding author’s e-mail: [email protected])
Received: 12 November 2025, Revised: 10 December 2025, Accepted: 17 December 2025, Published: 20 February 2026
Abstract
Cancer remains a significant global health burden due to high rates of morbidity, mortality, and disease recurrence. Apoptosis has emerged as an essential therapeutic target in cancer, and natural antioxidants from Indonesian medicinal plants show promise as modulators of apoptotic pathways because of their relative safety and bioactivity. This study aimed to identify phytochemical candidates with potential for adjuvant cancer therapy by evaluating their interaction with key apoptosis-regulating proteins. Nine antioxidant compounds (chlorogenic acid, cinnamic acid, curcumin, eugenol, gallic acid, kaempferol, quercetin, rutin, and tanshinone) were subjected to molecular docking against ten proteins representing intrinsic and extrinsic apoptotic pathways using AutoDock Vina, with dasatinib as a reference inhibitor. Drug-likeness was assessed according to Lipinski’s rules, while pharmacokinetics and toxicity predictions were performed using ADMETLab 3.0 and ProTox-II. The analysis showed that several compounds, particularly chlorogenic acid, curcumin, and quercetin, exhibited strong binding affinities to apoptosis-regulating proteins and interacted with critical amino acid residues associated with pro-apoptotic signaling. While rutin and tanshinone also demonstrated favorable docking scores, their pharmacokinetic limitations and predicted toxicity reduced their therapeutic potential. The combined evaluation of molecular binding, drug-likeness, and safety profiles supports chlorogenic acid, quercetin, and curcumin as the most viable candidates for further investigation. These findings indicate that specific plant-derived antioxidants may contribute to apoptosis-induced cancer inhibition and offer a rational basis for selecting lead compounds for future in vitro and in vivo validation. Overall, this study provides a systematic in silico framework to prioritize natural compounds as potential adjuvant therapeutic agents targeting apoptosis in cancer.
Keywords: Molecular docking, Natural antioxidants, Apoptosis, Adjuvant cancer therapy
Introduction
Cancer remains one of the leading causes of mortality worldwide, responsible for approximately 10 million deaths in 2020, thereby posing a major global health challenge [1]. Among all malignancies, lung cancer is the most frequently diagnosed and the deadliest, followed by colorectal, liver, stomach, and breast cancers, which collectively account for a significant proportion of cancer morbidity and mortality [1-3]. The economic burden of cancer treatment is
equally daunting, with global costs reaching hundreds of billions of dollars annually. This financial impact places substantial pressure on healthcare systems worldwide, particularly in low- and middle-income countries [2].
Although current treatment modalities, ranging from surgery, chemotherapy, and radiotherapy to immunotherapy and targeted molecular approaches, have improved patient outcomes, their overall effectiveness remains limited due to adverse side effects, restricted selectivity, and the persistent challenge of tumor recurrence [4,5]. High relapse rates in cancers such as colorectal, lung, and breast further highlight the urgent need for innovative therapeutic strategies capable of eradicating residual malignant cells [1]. Within this context, apoptosis, or programmed cell death, has emerged as a promising therapeutic target. Apoptosis plays a central role in maintaining tissue homeostasis and suppressing tumor progression, and its dysregulation not only facilitates unchecked proliferation but also contributes to chemoresistance [6,7]. Apoptotic signaling is primarily governed by two canonical pathways: The intrinsic (mitochondrial-mediated) and extrinsic (death receptor-mediated) cascades, both converging on caspase activation that orchestrates cellular dismantling [8,9]. Beyond these, alternative mechanisms such as necroptosis, ferroptosis, and immunogenic cell death are increasingly investigated to overcome apoptotic resistance and enhance therapeutic efficacy [6]. Thus, a deeper understanding of apoptosis and its molecular regulators remains vital for developing novel cancer treatments with greater selectivity and reduced toxicity [10,11].
In pursuit of this goal, plant-derived natural compounds have gained increasing attention for their ability to selectively modulate apoptotic pathways in cancer cells [12,13]. These bioactive agents are valuable for their structural diversity, favorable safety profiles, and multifaceted biological activities. Several antioxidants, such as curcumin, resveratrol, quercetin, and chlorogenic acid, have promoted apoptosis in various cancer models while sparing normal cells [13]. Significantly, resistance to conventional therapies is often associated with oxidative stress and defective apoptosis, making antioxidant-rich phytochemicals promising candidates to restore cell death mechanisms [14].
Indonesia, recognized as one of the world’s most biodiverse countries, provides a vast reservoir of natural products for drug discovery. With approximately 50,000 plant species, 7,500 of which contribute to traditional medicine, the nation harbors an immense pharmacopeia of bioactive compounds with untapped therapeutic potential [15,16]. Medicinal plants such as turmeric (Curcuma longa), ginger (Zingiber officinale), and temulawak (Curcuma xanthorrhiza) have long been applied for their anti-inflammatory and antioxidant effects in Indonesian traditional medicine [17,18]. Many of these species are rich in secondary metabolites, including alkaloids, flavonoids, phenolics, saponins, and curcuminoids, which have demonstrated the capacity to modulate oxidative stress and apoptosis-related signaling pathways [19,20]. Bridging ethnomedicinal knowledge with molecular approaches may accelerate the discovery of novel anticancer agents [21,22]. To systematically evaluate these compounds, in silico methods such as molecular docking have become indispensable tools in modern drug discovery. Docking enables rapid and cost-effective screening of ligands against molecular targets, predicting binding affinities, identifying critical interacting residues, and prioritizing candidates for further validation [15,23]. Platforms such as AutoDock provide reliable computational insights that, when combined with phytochemical screening, significantly enhance the identification of promising drug-like molecules [12]. Moreover, integrating molecular docking with ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiling offers a comprehensive strategy to evaluate both efficacy and pharmacokinetic feasibility, thereby increasing translational potential.
This study aims to investigate the molecular interactions between antioxidant compounds from selected Indonesian medicinal plants and apoptosis-regulating proteins using molecular docking, complemented by ADMET profiling. Combining ethnopharmacological resources with computational approaches, the research seeks to identify phytochemicals with significant anticancer potential and provide a rational foundation for their development as adjuvant therapeutic agents.
Materials and methods
Ligand selection
In this study, we selected nine antioxidant compounds which are chlorogenic acid (PubChem CID: 1794427), cinnamic acid (PubChem CID: 444539), curcumin (PubChem CID: 969516), eugenol (PubChem CID: 3314), gallic acid (PubChem CID: 370), kaempferol (PubChem CID: 5280863), quercetin (PubChem CID: 5280343), rutin (PubChem CID: 5280805), and tanshinone (PubChem CID: 164676) as ligands for molecular docking analysis. These compounds were selected based on their status as major constituents of inexpensive herbal plants, widely available in local Indonesian markets, and long utilized in traditional medicine. Furthermore, these antioxidants are commonly consumed in daily diets through spices, herbs, or vegetables, making them culturally and nutritionally relevant. Their ease of isolation and abundant availability also ensure sufficient supply for further pharmacological investigation [23]. Figure 1 presents a two-dimensional structural overview.
Receptor preparation
This study selected apoptosis as the central therapeutic mechanism due to its critical role in regulating programmed cell death and its relevance in overcoming cancer cell resistance. The investigation focuses on key molecular components involved in both the extrinsic (death receptor-mediated) and intrinsic (mitochondria-mediated) apoptotic pathways, particularly TNFR1, Fas Receptor, Bcl-2 family protein, caspase-9, caspase-8, caspase-3 and endonuclease G. These targets were chosen because they represent crucial nodes in the initiation and execution of apoptosis: TNFR1 and the Fas Receptor initiates death signaling via death signaling complex (DISC) formation, caspase-8 activates downstream caspases (including caspase-9 and caspase-3) and connects extrinsic to intrinsic pathways through Bcl-2 family protein cleavage, and endonuclease G mediates DNA fragmentation following mitochondrial outer membrane leakage [24]. The study aims to identify phytochemicals capable of inducing apoptosis selectively in cancer cells by targeting these proteins, providing a mechanistic basis for developing more effective and less toxic adjuvant anticancer therapies.
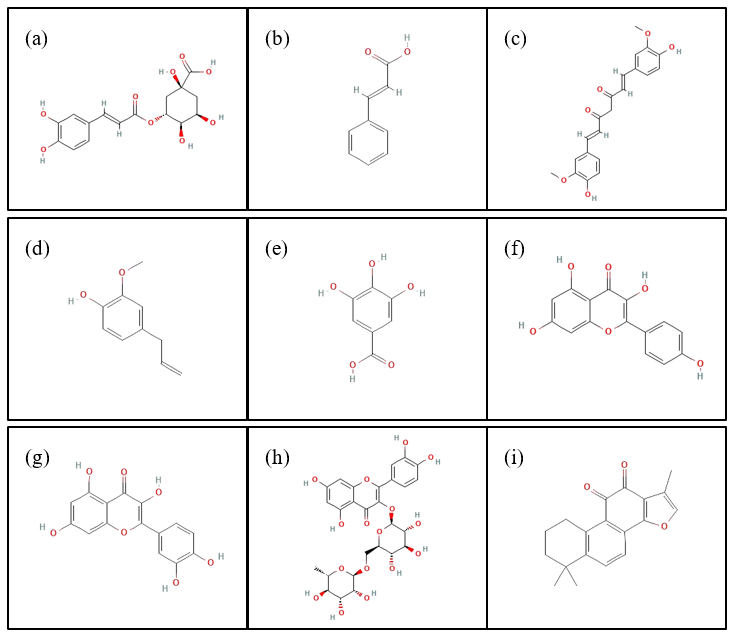
Figure 1 Two-dimensional structures of antioxidant compounds from Indonesian herbal plants: (a) chlorogenic acid, (b) cinnamic acid, (c) curcumin, (d) eugenol, (e) gallic acid, (f) kaempferol, (g) quercetin, (h) rutin, and (i) tanshinone.
The target proteins prepared for molecular docking included TNFR1 (PDB ID: 1NCF; resolution: 2.25 Å), Bid (PDB ID: 2BID), Bad (PDB ID: 2BZW chain A; resolution: 2.30 Å), Bax (PDB ID: 4BD6 chain A; resolution: 2.49 Å), Bak (PDB ID: 5FMI; resolution: 1.49 Å), FasR (PDB ID: 3EZQ chain A; resolution: 2.73 Å), EndoG (PDB ID: 6NJU chain D; resolution: 2.35 Å), caspase-8 (PDB ID: 7JKQ chain B; resolution: 3.30 Å), caspase-9 (PDB ID: 1NW9 chain B; resolution: 2.40 Å), and caspase-3 (PDB ID: 3H0E chain A and B; resolution: 2.00 Å), all retrieved from the Protein Data Bank (PDB, https://www.rcsb.org/). Before docking, AutoDockTools optimized the receptors by removing water molecules, detaching native ligands, eliminating non-standard residues, and adding polar hydrogens and Gasteiger charges [25].
Binding site prediction
The prepared selected receptors are then predicted for their active site using CASTpFold [26]. The PDB file of the cleaned-up protein is uploaded to the server, and the results are downloaded to be thoroughly analysed in Chimera 1.19 [27]. The selected active sites have the highest molecular surface volume and pocket area within the range of binding site residues, determined as the binding location.
ADMET prediction
We obtained 2D structures in .sdf format and SMILES strings from PubChem database (http://pubchem.ncbi.nlm.nih.gov) and input it to ADMETLab 3.0 (https://admetlab3.scbdd.com/) to perform assessment for the compoundsʼ Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profile. Descriptors such as the number of hydrogen bond donors and acceptors, molecular weight, bioavailability, and compliance with Lipinskiʼs Rule of Five guided the pharmacokinetic evaluation. The ProTox-II (https://tox.charite.de/protox3/) server generated toxicity predictions, following classifications aligned with the Globally Harmonised System (GHS).
Molecular docking simulation
Ligand preparation employed BIOVIA Discovery Studio (https://discover.3ds.com/discovery-studio-visualizer-download). Ligand structures originated from the PubChem database (http://pubchem.ncbi.nlm.nih.gov), and PubChem stored them in Standard Data Format (SDF) for further analysis. Open Babel performed energy minimization of the ligands [28]. Molecular docking simulations evaluated the binding of nine antioxidant compounds from Indonesian medicinal plants to ten apoptosis-regulating proteins. Docking also included a comparative analysis with the reference drug Dasatinib (PubChem CID: 3062316), a leukemia therapy agent [29]. AutoDock Vina conducted the docking runs, while Discovery Studio Visualizer displayed the results [30]. We used exhaustiveness value of 8 and num modes of 8 as our docking parameters. All receptors received a grid box of 60×60×60 with 0.375 Å spacing.
Results and discussion
Binding site prediction
Binding site prediction was performed for each receptor using the CASTpFold server to identify potential ligand-binding pockets based on surface topology and geometric parameters. This step was essential to ensure that docking simulations were carried out within the most biologically relevant cavities, thereby improving the reliability of the predicted interactions. Table 1 presents the identified binding sites and their structural characteristics.
Table 1 Active site mapping and grid box coordinates for molecular docking.
No |
Protein |
PDB ID |
Active site visualization |
Center Ref. Grid (Å) Coordinate |
1 |
TNFR1 |
1NCF |
|
X: 16; Y: 14; Z: 20 |
2 |
Bid |
2BID |
|
X: ‒13; Y: 0; Z: ‒2 |
3 |
Bad |
2BZWa |
|
X: 58; Y: 50; Z: 5 |
4 |
Bax |
4BD6a |
|
X: ‒2; Y: ‒26; Z: ‒15 |
5 |
Bak |
5FMI |
|
X: 0; Y: 7; Z: 24 |
6 |
FasR |
3EZQa |
|
X: ‒80; Y: 24; Z: ‒43 |
7 |
Endonuclease G |
6NJUd |
|
X: ‒15; Y: 39; Z: 30 |
8 |
Caspase-8 |
7JKQb |
|
X: 169; Y: 160; Z: 218 |
9 |
Caspase-9 |
1NW9b |
|
X: 51; Y: 10; Z: 90 |
10 |
Caspase-3 |
3H0E |
|
X: 25; Y: 47; Z: 12 |
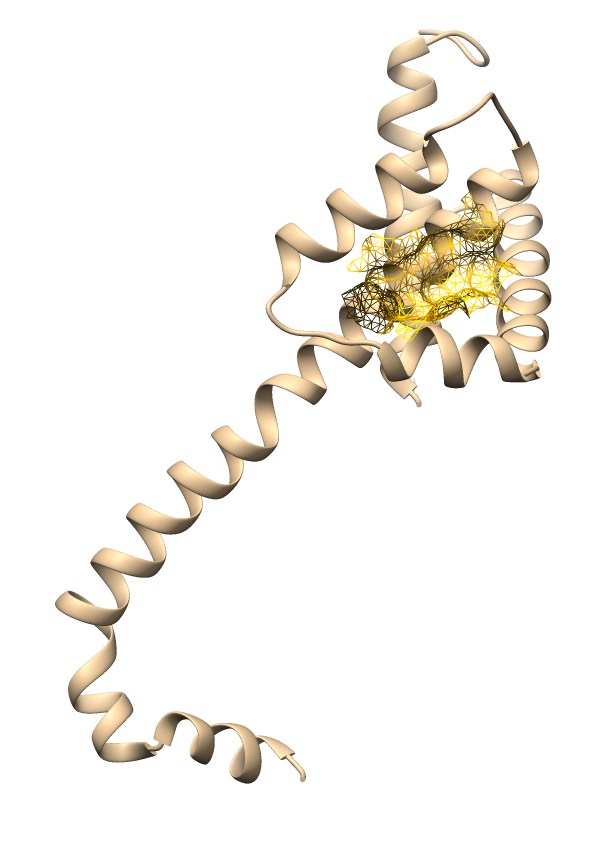
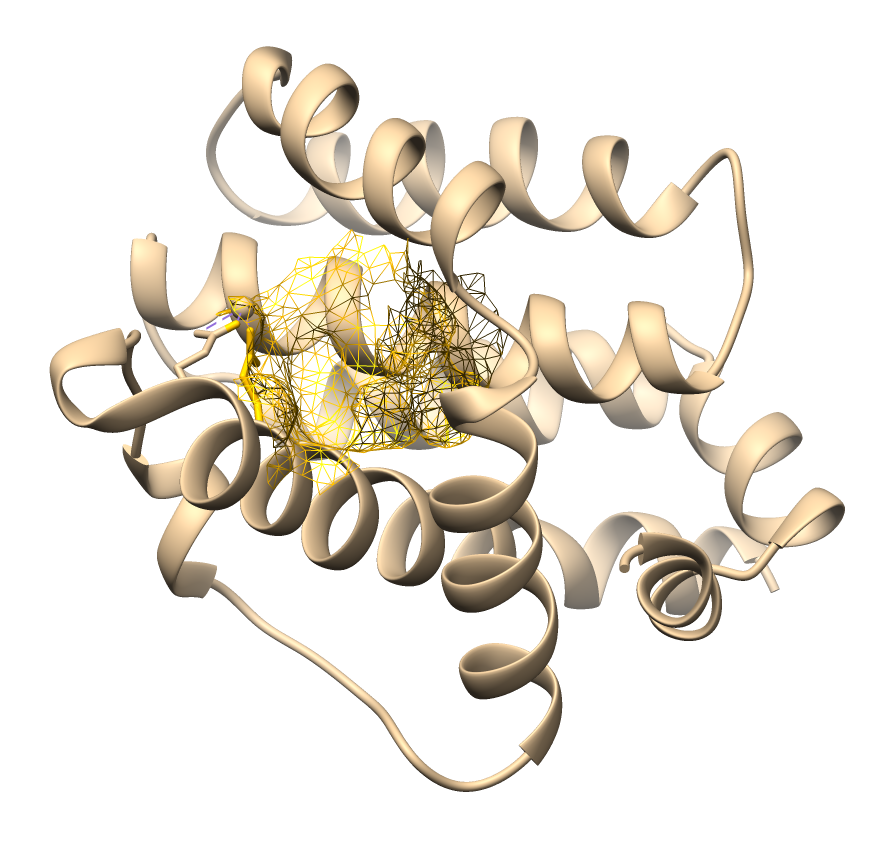
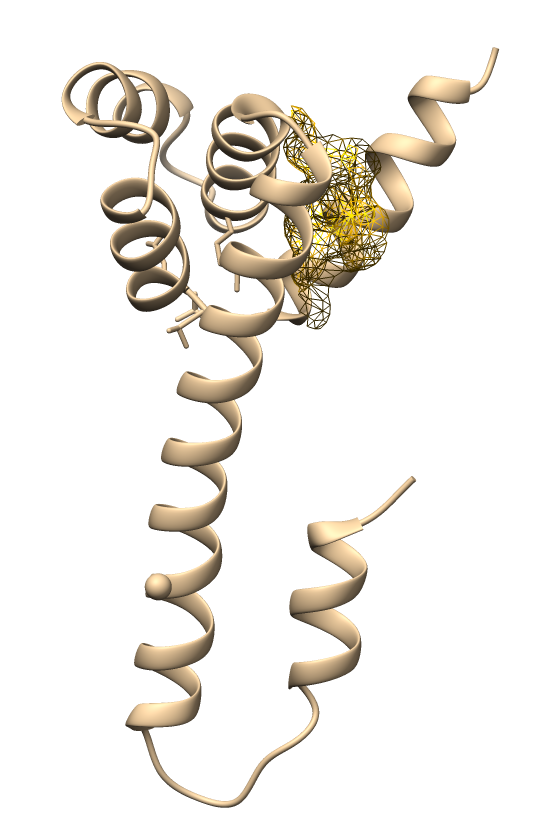
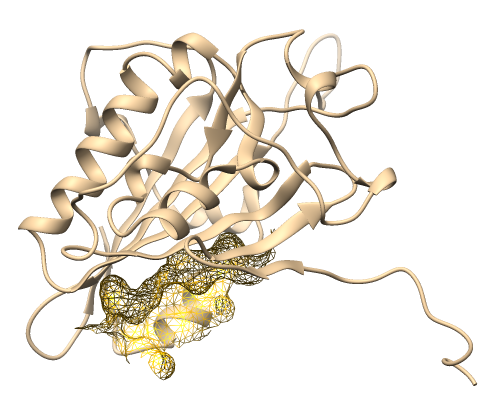
The predicted binding pockets correspond to key functional domains of apoptotic-regulating proteins. These results provide a reliable structural basis for subsequent docking simulations and ensure the assessment of ligand–receptor interactions within biologically relevant sites.
Drug-likeness evaluation
Assessing the drug-likeness of compounds is a vital preliminary phase in drug development, as it offers critical insights into their potential efficacy as therapeutic agents [31]. Evaluating the physicochemical properties of these compounds against established criteria, such as Lipinski's Rule of Five, facilitates the identification of favorable pharmacokinetic profiles. Table 2 presents the physicochemical analysis of the antioxidants and their compliance with Lipinskiʼs criteria.
Table 2 Predicted physicochemical and drug-likeness properties of antioxidant compounds based on Lipinski’s rule of five.
No |
Ligand |
Lipinski’s rule of five |
|||
MW |
logP ≤ 5 |
nHA |
nHD |
||
1 |
Chlorogenic Acid |
354.1 |
1.036 |
9.0 |
6.0* |
2 |
Cinnamic Acid |
148.05 |
2.619 |
2.0 |
1.0 |
3 |
Curcumin |
368.13 |
2.147 |
6.0 |
2.0 |
4 |
Eugenol |
164.06 |
2.321 |
2.0 |
1.0 |
5 |
Gallic Acid |
170.02 |
0.692 |
5.0 |
4.0 |
6 |
Kaempferol |
286.05 |
1.965 |
6.0 |
4.0 |
7 |
Quercetin |
302.04 |
1.448 |
7.0 |
5.0 |
8 |
Rutin |
610.15* |
0.986 |
16.0* |
10.0* |
9 |
Tanshinone |
294.13 |
3.646 |
3.0 |
0.0 |
Nearly all of the tested antioxidant compounds complied with Lipinskiʼs rule of five, which posits that orally active drug-like molecules typically possess a molecular weight ≤ 500 Da, logP ≤ 5, ≤ 5 hydrogen bond donors (nHD), and ≤ 10 hydrogen bond acceptors (nHA) [32]. Seven of the nine compounds, including cinnamic acid, curcumin, eugenol, gallic acid, kaempferol, quercetin, and tanshinone, satisfied all criteria, highlighting their favorable physicochemical properties and greater likelihood of exhibiting acceptable oral bioavailability.
Two compounds deviate from these criteria. Rutin exhibited three violations: a molecular weight of 610.15 Da, 16 hydrogen bond acceptors, and 10 hydrogen bond donors, which can significantly impair membrane permeability and intestinal absorption. These properties are consistent with pharmacokinetic studies reporting Rutin’s poor oral bioavailability, attributed to its low solubility, poor stability, and limited permeability across the intestinal epithelium due to its bulky sugar moiety [32,33]. Despite these limitations, Rutin retains biological relevance because intestinal microbiota can hydrolyze it into aglycone metabolites such as quercetin, which remain bioactive and contribute to its pharmacological effects [35]. Chlorogenic acid displayed only one deviation (nHD = 6), slightly above the donor threshold, which may modestly limit membrane diffusion efficiency. However, its relatively low molecular weight (354.1 Da) and favorable logP suggest it still holds oral delivery potential [32].
From a broader perspective, compliance with Lipinskiʼs criteria reinforces the potential of these phytochemicals to progress as orally available therapeutic candidates. Still, the rule should serve as a guiding framework rather than an absolute standard. Natural products frequently deviate from Lipinskiʼs thresholds yet remain pharmacologically active. For example, although Rutin have exceed the molecular weight limit of Lipinski’s Rule, it may still be developed as a therapeutic agent when delivered intravenously, bypassing the oral absorption barrier and broadening its translational potential in anticancer adjuvant therapy [36].
ADMET prediction
In oncology drug discovery, in silico approaches such as ADMET prediction and molecular docking are widely applied to identify novel inhibitors with favorable pharmacokinetic and pharmacodynamic properties. ADMET evaluation, which encompasses absorption, distribution, metabolism, excretion, and toxicity, represents a critical step in early drug discovery for predicting the in vivo behavior of candidate compounds. It provides insight into oral bioavailability, membrane permeability, metabolic stability, and potential drug-drug interactions, thereby complementing molecular docking analyses and strengthening the interpretation of docking outcomes [37]. This study subjected nine antioxidant compounds to ADMET profiling to evaluate their pharmacokinetic suitability as therapeutic candidates. Table 3 summarizes the predicted parameters of ADMET.
Table 3 Predicted ADMET properties of antioxidant compounds.
No |
Ligand |
Human Intestinal Absorption (HIA) |
F30% |
Caco-2 Permeability |
BBB Permeant |
CYP2D6 Substrate |
CYP2D6 Inhibitor |
CYP3A4 Substrate |
CYP3A4 Inhibitor |
T½ |
1 |
Chlorogenic Acid |
0.106 |
0.996 |
‒6.426 |
0 |
0 |
0 |
0 |
0 |
2.758 |
2 |
Cinnamic Acid |
0.095 |
0.89 |
‒4.441 |
0.025 |
0.086 |
0.006 |
0 |
0 |
1.882 |
3 |
Curcumin |
0.017 |
0.479 |
‒5.471 |
0.023 |
0.889 |
0.018 |
0 |
0.014 |
1.04 |
4 |
Eugenol |
0.035 |
0.985 |
‒4.57 |
0.106 |
0.999 |
0.342 |
0 |
0.998 |
1.464 |
5 |
Gallic Acid |
0.017 |
0.355 |
‒5.604 |
0.002 |
0 |
0 |
0 |
0.002 |
2.2 |
6 |
Kaempferol |
0.015 |
0.716 |
‒5.969 |
0.001 |
0.995 |
0.005 |
0.002 |
0.975 |
1.388 |
7 |
Quercetin |
0.134 |
0.991 |
‒6.177 |
0 |
0.978 |
0 |
0 |
0.937 |
1.586 |
8 |
Rutin |
0.64 |
1 |
‒6.547 |
0 |
0 |
0 |
0 |
0.029 |
4.616 |
9 |
Tanshinone |
0.763 |
0.664 |
‒4.612 |
0 |
0.002 |
0.995 |
0.644 |
0.849 |
0.499 |
The ADMET analysis revealed distinct pharmacokinetic and metabolic properties among the nine antioxidant ligands examined. Human intestinal absorption (HIA) is one of critical determinant of oral bioavailability. It was evaluated using a 30% threshold, where compounds with HIA ≥ 30% were classified as having good absorption and those with HIA < 30% as having poor absorption, and based on this classification, rutin and tanshinone exhibited poor absorption potential, corresponding to an HIA rate of < 30%. In contrast, curcumin and gallic acid demonstrated parameters consistent with higher oral bioavailability (HIA ≥ 30%), suggesting a more favorable pharmacokinetic profile [38,39].
Analysis of Caco-2 permeability, which reflects intestinal epithelial transport, identified cinnamic acid, eugenol, and tanshinone as the most permeable compounds, with values above the threshold of ‒ 5.15 log units [40]. These findings indicate that these ligands possess adequate membrane permeability, an important determinant for oral drug absorption. Despite this, all ligands consistently showed negative blood-brain barrier (BBB) penetration (category 0), indicating minimal central nervous system exposure, which may reduce the risk of neurotoxicity but also limits potential applications for neurological cancers [41].
Metabolic profiling indicated that several ligands may interact with cytochrome P450 enzymes. Specifically, curcumin, eugenol, kaempferol, and quercetin emerged as substrates of CYP2D6, highlighting their susceptibility to rapid metabolism. Furthermore, eugenol, kaempferol, quercetin, and tanshinone were identified as potential inhibitors of CYP3A4, suggesting a risk of drug-drug interactions with other therapeutics metabolized by this isoform. Such interactions warrant careful consideration in future drug development and clinical applications [42]. In terms of elimination, most compounds displayed moderate half-lives. However, tanshinone exhibited an exceptionally short half-life (0.499 h), which may compromise its therapeutic efficacy by limiting systemic exposure. Conversely, rutin showed the most extended half-life (4.616 h), but its poor intestinal absorption reduces its pharmacological relevance.
These findings suggest that among the nine antioxidants evaluated, curcumin and gallic acid present relatively favorable oral bioavailability, while cinnamic acid and eugenol exhibit superior permeability. However, potential liabilities include CYP-mediated interactions (particularly for eugenol, kaempferol, quercetin, and tanshinone) and poor absorption characteristics for rutin and tanshinone. These results underscore the importance of integrating ADMET profiling with docking analysis to prioritize candidates with strong binding affinities and acceptable pharmacokinetic properties.
Toxicity prediction
In addition to pharmacokinetic evaluation, toxicity assessment is essential in determining candidate compoundsʼ safety and therapeutic feasibility. Computational prediction of toxicological endpoints provides early insight into potential adverse effects, reducing the likelihood of late-stage failures in drug development [43]. In this study, we analysed nine antioxidant compounds for toxicity analysis, including the estimation of acute toxicity (LD50 values and toxicity classes) as well as organ-specific toxicities such as hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity. The results of these predictions appear in Tables 4 and 5.
Table 4 Predicted acute toxicity profiles of the antioxidant compounds.
No. |
Ligand |
Predicted LD50 (mg/kg) |
Predicted toxicity class |
Average similarity (%) |
Prediction accuracy (%) |
1 |
Chlorogenic Acid |
5,000 |
5 |
71.21 |
69.26 |
2 |
Cinnamic Acid |
2,500 |
5 |
100 |
100 |
3 |
Curcumin |
2,000 |
4 |
100 |
100 |
4 |
Eugenol |
1,930 |
4 |
100 |
100 |
5 |
Gallic Acid |
2,000 |
4 |
84.82 |
70.97 |
6 |
Kaempferol |
3,919 |
5 |
82.46 |
70.97 |
7 |
Quercetin |
159 |
3 |
100 |
100 |
8 |
Rutin |
5,000 |
5 |
100 |
100 |
9 |
Tanshinone |
1,655 |
4 |
48.84 |
54.26 |
Table 5 Predicted organ-specific toxicities of the antioxidant compounds.
No |
Ligand |
Organ toxicity |
||||
Hepatotoxicity (Acc. 93%) |
Carcinogenicity (Acc. 81%) |
Immunotoxicity (Acc. 75%) |
Mutagenicity (Acc. 84%) |
Cytotoxicity (Acc. 85%) |
||
1 |
Chlorogenic Acid |
Inactive |
Inactive |
Active |
Inactive |
Inactive |
2 |
Cinnamic Acid |
Active |
Inactive |
Inactive |
Inactive |
Inactive |
3 |
Curcumin |
Inactive |
Inactive |
Active |
Inactive |
Inactive |
4 |
Eugenol |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
5 |
Gallic Acid |
Inactive |
Active |
Inactive |
Inactive |
Inactive |
6 |
Kaempferol |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
7 |
Quercetin |
Inactive |
Active |
Inactive |
Active |
Inactive |
8 |
Rutin |
Inactive |
Inactive |
Active |
Inactive |
Inactive |
9 |
Tanshinone |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
The toxicological assessment of the nine antioxidant ligands revealed varying safety profiles, as indicated by their predicted LD50 values, toxicity classes, and organ-specific effects. In terms of acute toxicity, most ligands fell within toxicity classes 4 - 5, suggesting relatively low toxicity at standard exposure levels. Compounds such as chlorogenic acid, cinnamic acid, kaempferol, and rutin showed high LD50 values (≥ 2,500 mg/kg) and belonged to class 5, indicative of substances with limited acute toxicity. Curcumin, eugenol, gallic acid, and tanshinone occupied class 4, with LD50 values ranging between 1,655 and 2,000 mg/kg, reflecting moderate safety but warranting careful dose consideration [44,45]. Quercetin displayed the lowest LD50 (159 mg/kg), placing it in toxicity class 3, indicating its relatively higher acute toxicity compared to the other ligands. This finding aligns with previous studies that report quercetin’s higher toxicity due to its low LD50 value and potential carcinogenic and mutagenic effects [46].
Predicted organ toxicity provided further insights into potential risks. Chlorogenic acid, curcumin, and rutin demonstrated immunotoxic activity which may reflect their immunomodulatory properties at higher concentrations. Cinnamic acid showed hepatotoxic potential, raising caution regarding possible liver-related adverse effects upon prolonged use. Among the flavonoids, quercetin exhibited both carcinogenicity and mutagenicity, suggesting that despite its well-documented antioxidant benefits, its therapeutic window requires careful evaluation to minimize risks of genotoxicity. Similarly, gallic acid displayed carcinogenic potential, though other toxicity endpoints remained inactive. By contrast, kaempferol, eugenol, and tanshinone exhibited no activity across all assessed organ toxicity categories, underscoring their comparatively favorable safety profiles.
The toxicological profiling suggests that while most antioxidants investigated in this study exhibit acceptable safety margins, certain compounds, including quercetin and gallic acid, may pose risks related to mutagenicity and carcinogenicity. These findings emphasize the importance of dose optimization, structural modification, and comprehensive in vivo validation before advancing such compounds toward clinical applications. On the other hand, rutin, chlorogenic acid, and kaempferol stand out as relatively safe candidates given their high LD50 values and minimal predicted organ toxicity, reinforcing their suitability for further drug development efforts.
Molecular docking analysis
This analysis was performed to evaluate the binding affinities and interaction profiles of the nine selected antioxidant compounds against apoptosis-regulating proteins, including TNFR1, Bax, Bak, Bid, Bad, Fas receptor, Endonuclease G, Caspase-8, Caspase-9, and Caspase-3. These proteins represent critical nodes within intrinsic and extrinsic apoptotic pathways, making them valuable targets for anticancer drug discovery. The docking simulations provided insights into the potential mechanisms by which natural antioxidants may modulate apoptotic signaling by stabilizing pro-apoptotic proteins or interfering with anti-apoptotic regulators. Binding energy values (kcal/mol) were used as a primary indicator of ligand-protein affinity, complemented by an analysis of hydrogen bonding, hydrophobic contacts, and π-type interactions with key amino acid residues [47,48]. Dasatinib, a clinically used small-molecule kinase inhibitor, was the reference compound to validate docking accuracy and allow comparative evaluation [49]. Collectively, this approach identifies antioxidant ligands with strong binding potential and favorable interaction networks, thereby supporting their candidacy as modulators of apoptosis in cancer therapy. The results of the interaction and energy evaluation between molecular bonds are presented in Table 6.
Table 6 Molecular docking interactions and binding energies of antioxidant ligands.
Molecular bond interaction |
Chlorogenic acid |
Cinnamic acid |
Curcumin |
Eugenol |
Gallic acid |
Kaempferol |
Quercetin |
Rutin |
Tanshinone |
Dasatinib |
TNFR1 |
||||||||||
H-Bond |
HIS34 |
ALA62 |
LYS35 |
- |
HIS34 |
LYS35 |
HIS34 |
ASP91 |
GLU64 |
HIS34 |
π- Sigma π-Alkyl |
- |
LYS35 |
PHE60 |
HIS34 |
LYS35 |
LYS35 |
SER63 |
- |
- |
LYS35 |
π-Cation π-Anion π-Sulphur |
GLU64 |
GLU64 |
- |
LYS35 |
- |
GLU64 |
- |
LYS35 |
- |
LYS35 |
Binding Energy (Kcal/mol) |
‒6.7 |
‒5.3 |
‒6 |
‒5 |
‒5.6 |
‒6.3 |
‒6.7 |
‒6.8 |
‒6.7 |
‒6.4 |
Bid |
||||||||||
H-Bond |
GLN60 |
THR61 |
GLN60 |
GLN60 |
GLU54 |
TRP53 |
GLN60 |
GLN60 |
THR61 |
TRP53 |
π- Sigma π-Alkyl |
- |
TYR56 |
TYR56 |
TYR56 |
- |
- |
TYR56 |
- |
TYR56 |
- |
π-Cation π-Anion π-Sulphur |
GLU54 |
- |
- |
- |
- |
- |
- |
- |
- |
GLU54 |
Binding Energy (Kcal/mol) |
‒5.5 |
‒4.2 |
‒4.8 |
‒4 |
‒4.2 |
‒4.9 |
‒5.1 |
‒5.9 |
‒6.2 |
‒5.7 |
Bad |
||||||||||
H-Bond |
PHE105 |
- |
ARG102 |
PHE105 |
GLU153 |
ALA149 |
GLU98 |
LYS16 |
- |
ASP156 |
π- Sigma π-Alkyl |
LEU99 |
PHE105 |
- |
LYS16 |
ALA149 |
- |
ALA149 |
VAL152 |
LEU99 |
VAL152 |
π-Cation π-Anion π-Sulphur |
- |
- |
- |
- |
- |
- |
- |
LYS20 |
- |
LYS20 |
Binding Energy (Kcal/mol) |
‒7 |
‒5.2 |
‒7 |
‒5.3 |
‒5.3 |
‒6.6 |
‒6.6 |
‒8.2 |
‒8.3 |
‒8 |
Bax |
||||||||||
H-Bond |
ALA112 |
- |
ILE80 |
- |
ILE80 |
- |
- |
MET79 |
- |
MET99 |
π- Sigma π-Alkyl |
LEU76 |
LEU76 |
ALA112 |
MET99 |
VAL95 |
VAL95 |
LEU76 |
VAL95 |
LEU76 |
LEU76 |
π-Cation π-Anion π-Sulphur |
MET99 |
MET99 |
- |
- |
- |
MET99 |
MET99 |
- |
- |
ASP102 |
Binding Energy (Kcal/mol) |
‒6.2 |
‒5.4 |
‒6.8 |
‒5.3 |
‒4.3 |
‒6.6 |
‒6.6 |
‒7.3 |
‒7.7 |
‒7 |
Bak |
||||||||||
H-Bond |
GLN94 |
- |
ARG137 |
GLY82 |
ASP83 |
SER91 |
GLN94 |
SER91 |
ARG42 |
ARG137 |
π- Sigma π-Alkyl |
ARG87 |
ARG87 |
ALA49 |
ARG87 |
GLN45 |
ARG87 |
ALA49 |
GLN45 |
ARG87 |
ALA49 |
π-Cation π-Anion π-Sulphur |
ASP90 |
- |
GLU50 |
ASP83 |
- |
- |
ASP90 |
- |
ASP
90 |
ASP90 |
Binding Energy (Kcal/mol) |
‒6.8 |
‒4.7 |
‒6.3 |
‒5 |
‒5.7 |
‒6.5 |
‒6.8 |
‒7.6 |
‒6.8 |
‒7.1 |
Fas Receptor |
||||||||||
H-Bond |
SER225 |
LYS287 |
HIS282 |
- |
SER225 |
HIS282 |
TYR232 |
SER225 |
- |
SER225 |
π- Sigma π-Alkyl |
LEU 229 |
TYR232 |
ASP228 |
ALA290 |
LEU229 |
TYR232 |
TYR291 |
TYR232 |
LEU229 |
LEU298 |
π-Cation π-Anion π-Sulphur |
- |
- |
- |
HIS282 |
- |
- |
HIS282 |
HIS282 |
- |
TYR291 |
Binding Energy (Kcal/mol) |
‒5.7 |
‒4.5 |
‒5.8 |
‒4.2 |
‒4 |
‒6 |
‒6 |
‒6.6 |
‒6.7 |
‒6.6 |
Endonuclease G |
||||||||||
H-Bond |
ARG95 |
ARG96 |
ALA66 |
ASN133 |
ALA102 |
ALA97 |
ALA101 |
ASN128 |
ASN133 |
ALA101 |
π- Sigma π-Alkyl |
ALA68 |
ALA97 |
PHE71 |
ALA101 |
ALA100 |
ALA101 |
ALA68 |
ALA63 |
ALA102 |
ALA97 |
π-Cation π-Anion π-Sulphur |
- |
|
- |
- |
- |
ASP94 |
- |
- |
- |
- |
Binding Energy (Kcal/mol) |
‒7.4 |
‒5.1 |
‒7.7 |
‒5.4 |
‒6 |
‒7.3 |
‒7.7 |
‒8.9 |
‒7.8 |
‒7 |
Caspase-8 |
||||||||||
H-Bond |
HIS326 |
HIS252 |
SER193, |
- |
ASP349 |
GLU366 |
SER372 |
HIS326 |
TYR323 |
HIS324 |
π- Sigma π-Alkyl |
- |
PHE287 |
ILE347 |
PHE287 |
THR195 |
PHE370 |
ILE347 |
ILE347 |
PHE287 |
LEU256 |
π-Cation π-Anion π-Sulphur |
PHE370 |
- |
HIS326 |
- |
- |
HIS326 |
HIS326 |
- |
- |
PHE287 |
Binding Energy (Kcal/mol) |
‒6.7 |
‒6 |
‒6.3 |
‒5.6 |
‒6.2 |
‒6.2 |
‒6.4 |
‒7.5 |
‒7.1 |
‒7.2 |
Caspase-9 |
||||||||||
H-Bond |
THR181 ARG178 GLY176 HIS237 CYS287 |
ARG180
CYS287 |
ARG178 |
- |
- |
GLY176 |
THR181 |
GLY288 |
THR181 |
CYS287 |
π- Sigma π-Alkyl |
- |
- |
PHE351 |
ARG180 |
ARG180 |
ARG180 |
PRO357 |
ARG178 |
PRO357 |
LEU177 |
π-Cation π-Anion π-Sulphur |
ARG180 |
- |
CYS287 |
CYS287 |
CYS287 |
- |
- |
- |
- |
HIS237 |
Binding Energy (Kcal/mol) |
‒6.7 |
‒6 |
‒6.6 |
‒5.1 |
‒5.2 |
‒6.1 |
‒6.3 |
‒7 |
‒7.3 |
‒6.8 |
Caspase-3 |
||||||||||
H-Bond |
ARG207 |
- |
SER63 |
ARG207 |
TRP206 |
GLU123 |
SER205 |
SER209 |
CYS163 |
ARG207 |
π- Sigma π-Alkyl |
- |
PHE256 |
- |
PHE256 |
- |
- |
TYR204 |
TYR204 |
TYR204 |
MET61 |
π-Cation π-Anion π-Sulphur |
- |
- |
- |
- |
- |
- |
ARG207 |
- |
- |
CYS163 |
Binding Energy (Kcal/mol) |
‒7.1 |
‒5.3 |
‒6.2 |
‒5.2 |
‒4.8 |
‒6.5 |
‒6.9 |
‒7.6 |
‒7.9 |
‒7.3 |
TNFR1
The tumor necrosis factor receptor 1 (TNFR1) is a pivotal mediator of apoptotic and inflammatory signaling, making it an attractive target in cancer therapy. Molecular docking analysis revealed that rutin exhibited the strongest binding affinity (‒6.8 kcal/mol), followed closely by quercetin, chlorogenic acid, and tanshinone (‒6.7 kcal/mol each). Curcumin and kaempferol also demonstrated moderate affinity (‒6.0 and ‒6.3 kcal/mol, respectively). Key binding interactions involved hydrogen bonds with residues such as LYS35, GLU64, and HIS34, underscoring their importance in ligand stabilization within the receptor pocket. Dasatinib, a reference inhibitor, displayed a binding energy of ‒6.4 kcal/mol and established a balanced network of hydrogen bonds and hydrophilic contacts, validating the docking protocol. Curcumin formed a diverse interaction profile, including hydrogen bonding with LYS35 and GLU64 and multiple π-type interactions with PHE60, ARG92, ASP93, ALA62, and HIS34. These combined electrostatic and hydrophobic interactions likely confer enhanced conformational stability despite its lower binding affinity than rutin. From a translational perspective, while rutin and quercetin demonstrated stronger affinities, their unfavorable pharmacokinetic properties, including poor absorption and bioavailability, may restrict therapeutic application. Conversely, curcumin, with a slightly weaker binding score but superior drug-likeness, may represent a more viable candidate for further development.
Bid
Bid (BH3-interacting domain death agonist) is a pro-apoptotic Bcl-2 family protein that links the extrinsic and intrinsic apoptotic pathways. Based on previous research, the key residue that binds the ligand in Bid is GLN60 [50]. Docking analysis indicated that rutin (‒5.9 kcal/mol) and tanshinone (‒6.2 kcal/mol) showed the strongest binding affinities, surpassing other antioxidants but remaining slightly lower compared to the control dasatinib (‒5.7 kcal/mol). Rutin exhibited multiple hydrogen bonds with GLN60, LEU59, ARG65, and π-alkyl stabilization with TYR56, suggesting favorable anchoring within the binding pocket. Curcumin (‒4.8 kcal/mol) formed limited interactions, primarily with GLN60, while cinnamic acid and gallic acid demonstrated the weakest binding (‒4.2 kcal/mol). Overall, rutin and tanshinone appear to be the most promising ligands for Bid, potentially enhancing Bid-mediated mitochondrial apoptosis through stable ligand-protein interactions. Although tanshinone binds more tightly to the receptors, it does not form hydrogen bonds with GLN60, which might reduce target specificity. Quercetin, curcumin, and eugenol showed a similar bonding pattern to GLN60 and TYR56, which is considered the key binding pattern to the Bid Receptor.
Bad
Bad, or also commonly known to be Bcl-2-associated death promoter, is a BH3-only pro-apoptotic protein that antagonizes anti-apoptotic Bcl-2 family members, promoting apoptosis. Docking analysis showed that tanshinone (‒ 8.3 kcal/mol) and rutin (‒8.2 kcal/mol) exhibited the strongest binding affinities, surpassing dasatinib (‒8.0 kcal/mol). Curcumin (‒7.0 kcal/mol) and chlorogenic acid (‒7.0 kcal/mol) followed with moderate affinities. The ligands consistently interacted with key residues, including GLU153, PHE105, GLU98, VAL152, and LYS20. These findings indicate that tanshinone and rutin possess strong potential to modulate Bad activity, thereby enhancing intrinsic apoptotic signalling.
Bax
Bax is a pro-apoptotic protein that is central to mitochondrial-mediated apoptosis by permeabilizing the outer mitochondrial membrane. Molecular docking against Bax resulted in tanshinone (‒7.7 kcal/mol), rutin (‒7.3 kcal/mol), and Curcumin (‒6.8 kcal/mol) as top ligands with highly favorable binding energy similar to dasatinib (‒7.0 kcal/mol). Residues that play important roles in the binding to Bax are ALA112, LEU76, MET99, and ILE80. Although tanshinone has a greater binding energy with Bax, it exhibits no hydrogen bonding to the protein. Despite slightly lower affinity, Curcumin exhibited multiple stabilizing interactions and binding patterns similar to dasatinib, making it a promising candidate for Bax receptor modulation.
Bak
Bak, a significant pro-apoptotic Bcl-2 family member, functions in synergy with Bax to mediate mitochondrial outer membrane permeabilization, a critical step in apoptosis. The binding energy from molecular docking toward the Bak receptor ranged from ‒4.7 to ‒7.6 kcal/mol. Rutin exhibited the strongest binding energy (‒7.6 kcal/mol) among the tested compounds, surpassing dasatinib. Quercetin (‒6.8 kcal/mol) and chlorogenic acid (‒6.8 kcal/mol) also displayed high affinities comparable to dasatinib. Curcumin also showed fine binding energy stabilized by multiple hydrogen bonds with residues similar to those of dasatinib, which are ARG137, GLN45, and ARG42. Although Kaempferol has a slightly better binding energy than curcumin, it does not exhibit the same binding model. Besides that, ASP83 and ALA49 also play important roles in the binding of ligands in the Bak receptor.
Fas receptor
The Fas receptor is an extrinsic apoptosis pathway regulator, and ligand binding can initiate caspase cascades. From the molecular docking results, the binding energies of the tested ligands ranged from ‒4.0 to ‒6.7 kcal/mol. Tanshinone (‒6.7 kcal/mol) showed the strongest binding affinities, followed closely by rutin (‒6.6 kcal/mol), similar to dasatinib (‒6.6 kcal/mol). Quercetin (‒6.0 kcal/mol), kaempferol (‒6.0 kcal/mol), curcumin (‒5.8 kcal/mol), and chlorogenic acid (‒5.7 kcal/mol) showed almost similar binding affinities, whereas cinnamic acid and eugenol demonstrated weaker affinities (< ‒4.5 kcal/mol). Several residues commonly involved in ligand interactions are TYR291, HIS282, TYR232, and SER225, suggesting these are critical binding sites on the Fas receptor. Although Tanshinone showed the strongest binding, it still lacks hydrogen bonding, which could make the conformation unstable. These findings indicate that rutin and Tanshinone may effectively stabilize Fas receptor conformations, although their therapeutic potential remains dependent on pharmacokinetic optimization.
Endonuclease G (EndoG)
Endonuclease G is a mitochondria-localized nuclease involved in caspase-independent apoptosis. Binding energy from the molecular docking toward Endonuclease G ranged from ‒5.1 to ‒8.9 kcal/mol. Rutin exhibited the strongest binding (‒8.9 kcal/mol), surpassing dasatinib. Rutin established multiple hydrogen bonds with ASN128, ALA97, and ARG106, alongside extensive hydrophobic contacts (PHE71, ALA101, ALA102). These interactions highlight rutin’s ability to anchor deeply into the EndoG binding pocket; otherwise, the pattern differs from dasatinib. Tanshinone, curcumin, and quercetin also demonstrated high affinities around ‒7.7 kcal/mol. The three with substantial binding stability and overlapping residue interactions with the reference drug emerge as a promising natural ligand of Endonuclease G-mediated apoptosis.
Caspase-8
Caspase-8 is a critical initiator caspase involved in the extrinsic apoptosis signalling pathway. Molecular docking results against the caspase-8 receptor resulted in binding energies ranging from ‒5.6 to ‒7.5 kcal/mol. Rutin exhibited the strongest binding affinity, followed by tanshinone, both of which have similar binding energy to Dasatinib. Chlorogenic acid, quercetin, and Curcumin also demonstrated favorable interactions toward caspase-8. The key residues in the caspase eight protein that bind to ligands are ILE347, HIS326, HIS324, TYR323, and PHE287. Compared with Dasatinib, rutin and tanshinone demonstrated nearly equivalent binding energies, suggesting their potential as potent natural caspase-8 modulators. While moderately strong, Curcumin displayed multiple hydrogen bonds and hydrophobic interactions overlapping with residues bound by Dasatinib (ILE347, PHE287, and HIS326), indicating a similar inhibitory mechanism despite a slightly weaker docking score.
Caspase-9
Caspase-9 is a crucial initiator in the intrinsic apoptotic pathway, activated via the apoptosome complex. It showed binding energies ranging from ‒5.1 to ‒7.3 kcal/mol, with tanshinone (‒7.3 kcal/mol) and rutin (‒7.0 kcal/mol) exhibiting the highest binding energy. Chlorogenic acid (‒6.7 kcal/mol) and curcumin (‒6.6 kcal/mol) showed similar binding energy with the reference drug dasatinib (‒6.8 kcal/mol). Comparative analysis demonstrated that tanshinone and rutin had stronger binding energies than dasatinib, which appear to be promising ligands for caspase-9 activation, supporting the role of natural antioxidants in promoting mitochondrial apoptosis. The residues commonly binding to ligands are PHE351, CYS287, HIS237, ARG180, and THR179.
Caspase-3
Caspase-3 is a critical executioner caspase responsible for the final stages of apoptosis. The molecular docking results showed binding energies ranged from ‒4.8 to ‒7.9 kcal/mol, with tanshinone (‒7.9 kcal/mol) and rutin (‒7.6 kcal/mol) exhibiting similar binding energies with dasatinib. Quercetin (‒6.9 kcal/mol), kaempferol (‒6.5 kcal/mol), and curcumin (‒6.2 kcal/mol) showed moderate binding energy, indicating favorable interaction with caspase-3. The commonly similar residues that bind caspase 3 to ligands are PHE256, ARG207, TRP206, TYR204, and CYS163.
Candidate evaluation in apoptotic pathways
Among the tested antioxidants, chlorogenic acid demonstrated relatively strong binding affinities with apoptosis-regulating proteins, underlining its potential as a modulator of apoptotic signaling. Lipinskiʼs evaluation showed hydrogen bond donor (nHD) parameter violation, which may limit its oral absorption and permeability [32]. Despite this, ADMET analysis indicated only two unfavorable pharmacokinetic scores, while toxicity profiling suggested no major safety concerns. This positions chlorogenic acid as a viable candidate for further exploration, especially if supported by formulation strategies aimed at improving its bioavailability. In order to mitigate this pharmacokinetic limitations, formulation approaches such as phospholipid-based nanophytovesicles, phytosome complexes, liposomal carriers, and chitosan nanoparticles have been shown to enhance chlorogenic acidʼs lipid solubility, intestinal permeability, and systemic exposure [51].
Curcumin also exhibited notable molecular docking interactions, forming multiple hydrogen bonds and non-covalent contacts with key residues that reinforce binding stability. Although ADMET evaluation flagged two unfavorable pharmacokinetic properties, the overall profile remains acceptable. Furthermore, curcumin maintained a relatively safe toxicological profile, supporting its suitability as a therapeutic scaffold. The well-documented limitation of curcuminʼs poor systemic bioavailability could be overcome through nanocarrier delivery, combination therapy, or structural optimization, thereby enhancing its therapeutic efficacy in apoptosis regulation [52].
Quercetin and rutin displayed strong binding affinities toward apoptotic proteins but presented significant limitations in drug-likeness and safety profiles. Quercetin was predicted to have a relatively low LD50 and showed positive alerts for carcinogenicity and mutagenicity, raising concerns about its potential risks in therapeutic applications despite its natural dietary abundance [44]. Conversely, rutin exhibited strong binding energy and failed three of Lipinskiʼs rule criteria due to its high molecular weight and excessive hydrogen bonding potential, suggesting poor permeability and absorption [32]. ADMET predictions further highlighted unfavorable pharmacokinetics, undermining its development as a drug-like molecule. Therefore, while quercetin and rutin are biochemically active, their clinical translation may be constrained without substantial chemical modification or restriction to adjunctive, nutraceutical applications.
Tanshinone represents another strong-binding compound, yet its pharmacokinetic and toxicological characteristics pose significant hurdles. ADMET profiling indicated poor absorption and a short half-life, limiting systemic exposure. Moreover, toxicity analysis revealed a low predicted LD50, suggesting reduced safety margins compared to other tested antioxidants. Despite its molecular potential, these unfavorable properties substantially reduce tanshinone’s feasibility as a direct therapeutic candidate. Strategies such as advanced drug delivery formulations may be required to harness its bioactivity while minimizing pharmacological and toxicological drawbacks [53].
In contrast to the compounds with stronger binding energies, cinnamic acid and gallic acid exhibited relatively weaker affinities toward apoptotic proteins. However, their pharmacokinetic evaluation revealed more favorable properties, with both compounds showing the least unfavorable ADMET scores. These results suggest that although their direct interaction strength with apoptosis-related targets may not be ideal, their pharmacokinetic stability and drug-likeness make them promising substrates for additional modification. Significantly, their smaller molecular size and favorable absorption potential could facilitate structural modification to improve binding interactions without substantially compromising pharmacokinetics, thus positioning them as promising lead molecules for rational drug design.
Meanwhile, kaempferol and eugenol demonstrated intermediate docking affinities and acceptable pharmacokinetic characteristics, with kaempferol showing susceptibility to CYP2D6 metabolism and eugenol acting as an inhibitor of CYP3A4. These features suggest potential drug-drug interactions that must be carefully considered in therapeutic contexts. Nevertheless, both compounds are widely consumed in dietary sources and are known to exert antioxidant and anti-inflammatory activities, supporting their biological relevance. Their moderate binding strength and relatively balanced ADMET profiles indicate that kaempferol and eugenol could serve as supportive therapeutic agents, potentially in synergistic formulations with stronger-binding molecules such as curcumin or chlorogenic acid.
Overall, the molecular docking analysis highlights that several antioxidant compounds, particularly chlorogenic acid, curcumin, quercetin, rutin, and tanshinone, exhibit strong binding affinities toward apoptosis-regulating proteins, suggesting potential roles in modulating apoptotic pathways. However, their pharmacokinetic and toxicological profiles vary considerably, with some compounds displaying limited drug-likeness or safety concerns. In contrast, cinnamic acid, gallic acid, kaempferol, and eugenol demonstrated more favorable ADMET characteristics despite lower docking affinities, indicating their promise as lead scaffolds for optimization. These findings collectively emphasize the importance of integrating docking results with pharmacokinetic and toxicological evaluations to identify antioxidant candidates with strong molecular interactions and feasible drug development potential.
Molecular interactions visualizations
This study focused on three compounds with consistently strong binding affinities to provide deeper insights into the molecular mechanisms underlying their pro-apoptotic potential. Two-dimensional and three-dimensional molecular docking visualizations were generated for chlorogenic acid (Figure 2), curcumin (Figure 3), and quercetin (Figure 4). Rutin and tanshinone were excluded from the visualization due to unfavorable ADMET profiles and poor drug-likeness scores. We selected ligand-protein interactions with binding energies lower than –6 kcal/mol, ensuring that the analysis emphasized biologically meaningful and energetically favorable complexes. These visualizations provide detailed insights into specific hydrogen bonds, hydrophobic contacts, and electrostatic interactions between the ligands and key amino acid residues within the binding pockets. Both docking energy values with interaction mapping analysis strengthens the biological relevance of the predicted binding modes. It supports the identification of potential lead compounds for future in vitro and in vivo validation.
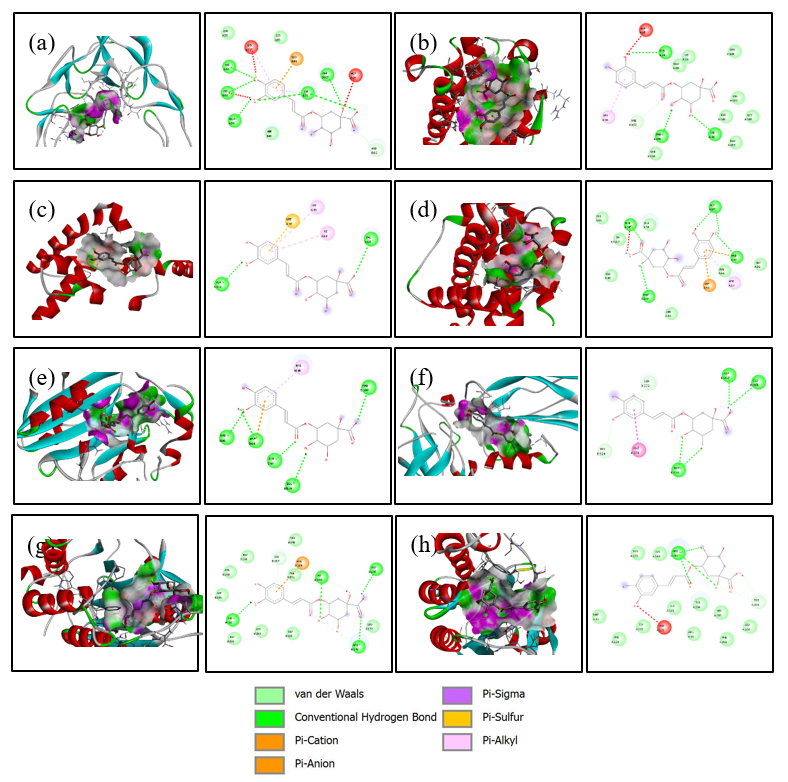
Figure 2 Visualization 3D and 2D of interactions of Chlorogenic Acid against the apoptotic-regulating protein. (a) TNFR1, (b) Bad, (c) Bax, (d) Bak, (e) Endonuclease G, (f) caspase-8, (g) caspase-9, and (h) caspase-3.
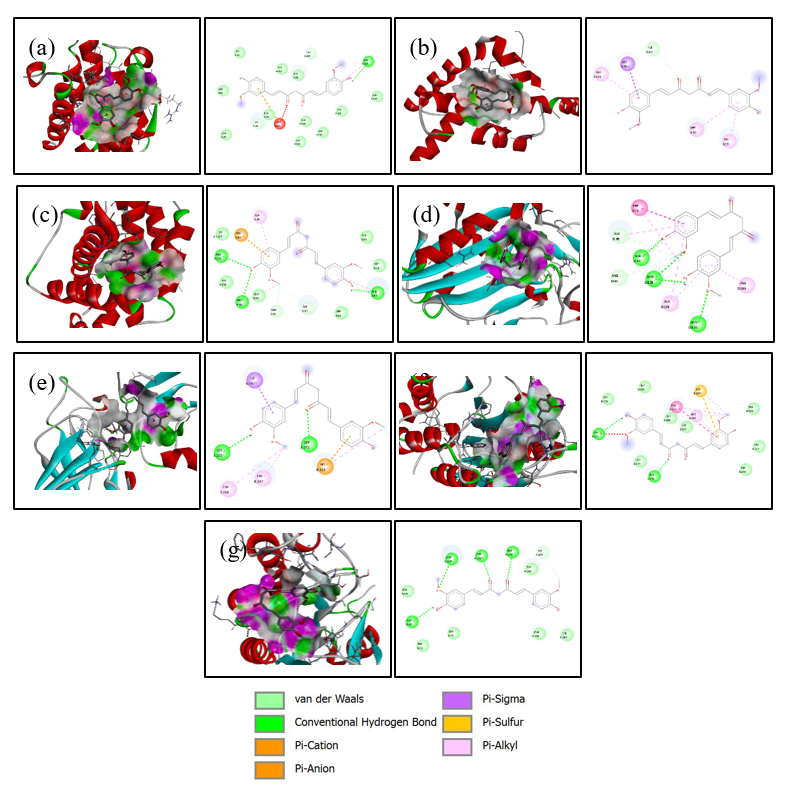
Figure 3 Visualization 3D and 2D of interactions of Curcumin against the apoptotic-regulating protein. (a) Bad, (b) Bax, (c) Bak, (d) Endonuclease G, (e) caspase-8, (f) caspase-9, and (g) caspase-3.
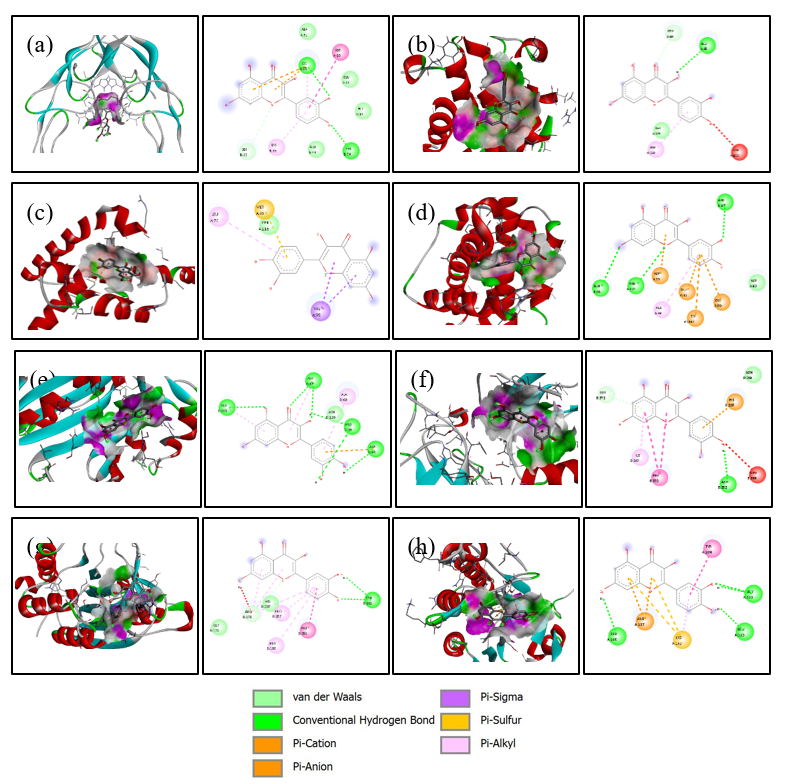
Figure 4 Visualization 3D and 2D of interactions of Quercetin against the apoptotic-regulating protein. (a) TNFR1, (b) Bad, (c) Bax, (d) Bak, (e) Endonuclease G, (f) caspase-8, (g) caspase-9, and (h) caspase-3.
Conclusions
This study comprehensively evaluated nine antioxidant compounds derived from Indonesian herbal plants through molecular docking, ADMET profiling, and toxicity prediction against key apoptotic-regulating proteins. This study revealed that several compounds, including chlorogenic acid, curcumin, quercetin, rutin, and tanshinone, exhibited strong binding affinities toward almost all apoptotic-regulating proteins, especially Bad, endonuclease G, caspase-8, and caspase-3 proteins. Suggesting their potential role in modulating apoptotic pathways. ADMET analysis indicated that most compounds possess favorable pharmacokinetic properties, although rutin and tanshinone showed limited absorption and bioavailability, kaempferol, and eugenol demonstrated possible CYP-mediated interactions. Toxicity predictions further confirmed that most compounds fell into safe toxicity classes with low risk of organ-specific toxicities. These findings highlight the therapeutic promise of selected natural antioxidants as potential lead compounds in cancer adjuvant therapy, while also underscoring the need for further in vitro and in vivo validation to confirm their efficacy and safety. In vitro analysis could be conducted to search for activity and efficacy by using enzyme assays for each receptor that is tested. Furthermore, in vivo analysis also could be used to evaluate the compounds efficacy in living organisms while also seeing the safety and toxicity of the compounds itself.
This study is still limited to the exploration of possible interactions between the antioxidant compound we used as ligand with many receptors that could contribute to cell apoptosis by using molecular docking simulations. There forewe didn’t conduct molecular dynamics simulations as it is not part of the study and it is the limitation of this study.
Future studies should focus on validating these in silico findings through in vitro and in vivo experiments to establish the biological relevance of the predicted interactions. Isolation and standardized extraction of the identified antioxidants from Indonesian herbal plants could provide a sustainable source for further pharmacological testing. Moreover, structural optimization and formulation approaches, such as nanoparticle-based delivery systems, may help overcome limitations in solubility, bioavailability, and metabolic stability observed in some compounds, particularly rutin and tanshinone. Integration of advanced computational techniques, including molecular dynamics simulations and quantitative structure-activity relationship (QSAR) modeling, would also enhance the prediction accuracy of ligand-protein interactions and pharmacokinetic behaviors. Ultimately, these efforts may accelerate the translation of natural antioxidants into novel adjuvant therapeutic agents targeting apoptotic-regulating proteins for cancer treatment.
Acknowledgements
The authors would like to thank the Faculty of Medicine, Universitas Negeri Semarang, for providing academic support and research facilities. We also sincerely appreciate our colleagues for their technical assistance and for supporting this study, particularly through the provision of specialized research software that enabled the completion of the computational analyses.
Conflict of interest
The authors declare no conflict of interest.
Declaration of Generative AI in Scientific Writing
The authors acknowledge the use of generative AI tools (e.g., QuillBot and ChatGPT by OpenAI) in the preparation of this manuscript, specifically for language editing and grammar correction. AI performed no content generation or data interpretation. The authors take full responsibility for the content and conclusions of this work.
CRediT Author Statement
Luqman Alwi: Conceptualization, Methodology, Investigation. Irwan Budiono: Validation, Data Curation, Visualization. Mahalul Azam: Formal analysis, Resources. Selamat Budijitno: Writing – Review & Editing, Supervision. Irena Intania: Writing – Original Draft, Funding acquisition. Matheus Prayoga: Software, Project administration.
References
[1] H Sung, J Ferlay, RL Siegel, M Laversanne, I Soerjomataram, A Jemal and F Bray. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians 2021; 71(3), 209-249.
[2] J Ferlay, M Colombet, I Soerjomataram, DM Parkin, M Piñeros, A Znaor and F Bray. Cancer statistics for the year 2020: An overview. International Journal of Cancer 2021; 149(4), 778-789.
[3] RL Siegel, KD Miller and A Jemal. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians 2016; 66(1), 7-30.
[4] Z Si, J Ying and Y Zhou. A study on the global burden of non-melanoma skin cancer from 1990 to 2019. Archives of Medical Science 2024; 20(6), 1902-1908.
[5] AN Hata, JA Engelman and AC Faber. The BCL2 family: Key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discovery 2015; 5(5), 475-487.
[6] F Pentimalli, S Grelli, ND Daniele, G Melino and I Amelio. Cell death pathologies: Targeting death pathways and the immune system for cancer therapy. Genes and Immunity 2019; 20, 539-554.
[7] CM Pfeffer and ATK Singh. Apoptosis: A target for anticancer therapy. International Journal of Molecular 2018; 19(2), 448.
[8] MN Messmer, AG Snyder and A Oberst. Comparing the effects of different cell death programs in tumor progression and immunotherapy. Cell Death & Differentiation 2019; 26(1), 115-129.
[9] S Baig, I Seevasant, J Mohamad, A Mukheem, HZ Huri and T Kamarul. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death & Disease 2016; 7(1), 2058.
[10] A Yaghoubi, M Ghojazadeh, S Abolhasani, H Alikhah and F Khaki-Khatibi. Correlation of serum levels of vitronectin, malondialdehyde and Hs-CRP with disease severity in coronary artery disease. Journal of Cardiovascular and Thoracic Research 2015; 7(3), 113-117.
[11] S Rajabi, M Maresca, AV Yumashev, R Choopani and H Hajimehdipoor. The most competent plant‐derived natural products for targeting apoptosis in cancer therapy. Biomolecules 2021; 11(4), 534.
[12] SR Lin, CH Chang, CF Hsu, MJ Tsai, H Cheng, MK Leong, PJ Sung, JC Chen and CF Weng. Natural compounds as potential adjuvants to cancer therapy: Preclinical evidence. British Journal of Pharmacology 2020; 177(6), 1409-1423.
[13] ML Madhuri and RR Reddy. In vitro evaluation and molecular docking analysis of potential anticancer compounds from Syzygium alternifolium. International Research Journal of Pure and Applied Chemistry 2022; 23(6), 1-8.
[14] JK Paul, M Azmal, ANMSNB Haque, OF Talukder, M Meem and A Ghosh. Phytochemical-mediated modulation of signaling pathways: A promising avenue for drug discovery. Advances in Redox Research 2024; 13, 100113.
[15] B Setiyono, MR Arif, QQ Aini, TH Soegianto, J Ohanna, RAF Gunawan and AP Rizkia. Identifikasi tanaman obat indonesia melalui citra daun menggunakan metode Convolutional Neural Network (CNN). Jurnal Teknologi Informasi dan Ilmu Komputer 2023; 10(2), 385-392.
[16] M Apriana, RMS Toni, MC Huda, ZM Kamal, R Khoerunnisa, Allahuddin, RA Septiani, SR Ash-Shidiqi and F Anggraeni. Pengobatan penyakit kolestrol dengan menggunakan ekstrak herbal di indonesia - a review. Jurnal Buana Farma 2022; 2(2), 12-32.
[17] MR Adiyasa and M Meiyanti. Pemanfaatan obat tradisional di Indonesia: Distribusi dan faktor demografis yang berpengaruh. Jurnal Biomedika dan Kesehatan 2021; 4(3), 130-138.
[18] E Emelda, R Nugraeni and K Damayanti. Review: Exploration of Indonesian herbal plants for antiinflammatory. INPHARNMED Journal (Indonesian Pharmacy and Natural Medicine Journal) 2023; 6(2), 58.
[19] YK Dewi and BA Riyandari. Potensi tanaman lokal sebagai tanaman obat dalam menghambat penyebaran COVID-19. Jurnal Pharmascience 2020; 7(2), 112-128.
[20] A Sarasati, MH Syahruddin, A Nuryanti, ID Ana, A Barlian, CH Wijaya, D Ratnadewi, TDK Wungu and H Takemori. Plant-derived exosome-like nanoparticles for biomedical applications and regenerative therapy. Biomedicines 2023; 11(4), 1053.
[21] M Alfarizi. Pengobatan komplementer alternatif lokal dan potensinya di Indonesia dalam perspektif kesehatan dan ekonomi: Kajian literatur sistematik. Salus Cultura: Jurnal Pembangunan Manusia dan Kebudayaan 2022, 2(2), 138-150.
[22] M Devi, D Dahiya, N Sharma, A Soni, C Kumari and Yamini. In silico molecular docking study of some novel chalcone derivatives as anticancer agents. Journal of Pharmaceutical Research International 2023; 35(22), 54-65.
[23] T Estiasih, JM Maligan, JE Witoyo, AAH Mu’Alim, K Ahmadi, T Mahatmanto and E Zubaidah. Indonesian traditional herbal drinks: Diversity, processing, and health benefits. Journal of Ethnic Foods 2025; 12, 7.
[24] V Shoshan-Barmatz, T Arif and A Shteinfer-Kuzmine. Apoptotic proteins with non-apoptotic activity: Expression and function in cancer. Apoptosis 2023; 28(5-6), 730-753.
[25] GM Morris, H Ruth, W Lindstrom, MF Sanner, RK Belew, DS Goodsell and AJ Olson. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry 2009; 30(16), 2785-2791.
[26] B Ye, W Tian, B Wang and J Liang. CASTpFold: Computed atlas of surface topography of the universe of protein folds. Nucleic Acids Research 2024; 52(W1), W194-W199.
[27] EF Pettersen, TD Goddard, CC Huang, GS Couch, DM Greenblatt, EC Meng and TE Ferrin. UCSF Chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry 2004; 25(13), 1605-1612.
[28] NM O’Boyle, M Banck, CA James, C Morley, T Vandermeersch and GR Hutchison. Open babel: An open chemical toolbox. Journal of Cheminformatics 2011; 3, 33.
[29] PR Sankar, AB Sailu, MM Eswarudu, MN Satya, P Sreeja, P Roja and S Rijwana. Analytical methods for determination of different members of FDA approved tyrosine kinase inhibitors like Dasatinib, Lapatinib, Imatinib, Sorafenib, Nintedanib, Sunitinib and Pazopanib: A review. Journal of Pharmaceutical Sciences and Research 2021; 13(6), 313-318.
[30] O Trott and AJ Olson. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 2010; 31(2), 455-461.
[31] M Bitew, T Desalegn, TB Demissie, A Belayneh, M Endale and R Eswaramoorthy. Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. PLoS One 2021; 16(12), 0260853.
[32] CA Lipinski, F Lombardo, BW Dominy and PJ Feeney. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 2012; 23(1-3), 3-25.
[33] B Gullón, TA Lú-Chau, MT Moreira, JM Lema and G Eibes. Rutin: A review on extraction, identification and purification methods, biological activities and approaches to enhance its bioavailability. Trends in Food Science & Technology 2017; 67, 220-235.
[34] SR Akash, A Tabassum, LM Aditee, A Rahman, MI Hossain, MA Hannan and MJ Uddin. Pharmacological insight of rutin as a potential candidate against peptic ulcer. Biomedicine & Pharmacotherapy 2024; 177, 116961.
[35] J Terao. Potential role of quercetin glycosides as anti-atherosclerotic food-derived factors for human health. Antioxidants 2023; 12(2), 258.
[36] T Przybylski, J Czerniel, J Dobrosielski and M Stawny. Flavonol technology: From the compounds’ chemistry to clinical research. Molecules 2025; 30(15), 3113.
[37] JPB Arango, DYM Rodriguez, SL Cruz and GT Ocampo. In silico evaluation of pharmacokinetic properties and molecular docking for the identification of potential anticancer compounds. Computational Biology and Chemistry 2026; 120(1), 108626.
[38] LF Araujo, CHS de Pinto and LF Motta. In silico pharmacokinetic and toxicological study of cinnamic acid analogues. Brazilian Journal of Development 2022; 8(12), 80800-80817.
[39] NN Wang, C Huang, J Dong, ZJ Yao, MF Zhu, ZK Deng, B Lv, AP Lu, AF Chenac and DS Cao. Predicting human intestinal absorption with modified random forest approach: A comprehensive evaluation of molecular representation, unbalanced data, and applicability domain issues. RSC Advances 2017; 7, 19007-19018.
[40] I Ahmad, AE Kuznetsov, AS Pirzada, KF Alsharif, M Daglia and H Khan. Computational pharmacology and computational chemistry of 4-hydroxyisoleucine: Physicochemical, pharmacokinetic, and DFT-based approaches. Frontiers in Chemistry 2023; 11, 1145974.
[41] R Kato, L Zhang, N Kinatukara, R Huang, A Asthana, C Weber, M Xia, X Xu and P Shah. Investigating blood–brain barrier penetration and neurotoxicity of natural products for central nervous system drug development. Scientific Reports 2025; 15, 7431.
[42] J Zhai, VH Man, B Ji, L Cai and J Wang. Comparison and summary of in silico prediction tools for CYP450-mediated drug metabolism. Drug Discovery Today 2023; 28(10), 103728.
[43] AMB Amorim, LF Piochi, AT Gaspar, AJ Preto, N Rosário-Ferreira and IS Moreira. Advancing drug safety in drug development: Bridging computational predictions for enhanced toxicity prediction. Chemical Research in Toxicology 2024; 37(6), 827-849.
[44] United Nations. Globally Harmonized System of Classification and Labelling of Chemicals (GHS). 9th ed. United Nations, New York and Geneva, 2021.
[45] CM Zwickl, JC Graham, RA Jolly, A Bassan, E Ahlberg, A Amberg, LT Anger, L Beilke, P Bellion, A Brigo, H Burleigh-Flayer, MTD Cronin, AA Devlin, T Fish, S Glowienke, K Gromek, AL Karmaus, R Kemper, S Kulkarni, ..., GJ Myatt. Principles and procedures for assessment of acute toxicity incorporating in silico methods. Computational Toxicology 2022; 24, 100237.
[46] R Chowdhury, MS Bhuia, A Islam Rakib, R Hasan, HDM Coutinho, IM Araújo, IRA de Menezes and MT Islam. Assessment of quercetin antiemetic properties: In vivo and in silico investigations on receptor binding affinity and synergistic effects. Plants 2023; 12(24), 4189.
[47] APD Nurhayati, A Rihandoko, A Fadlan, SS Ghaissani, N Jadid and E Setiawan. Anti-cancer potency by induced apoptosis by molecular docking P53, caspase, cyclin D1, cytotoxicity analysis and phagocytosis activity of trisindoline 1,3 and 4. Saudi Pharmaceutical Journal 2022; 30(9), 1345-1359.
[48] EM Terefe and A Ghosh. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of phytochemicals isolated from croton dichogamus against the HIV-1 reverse transcriptase. Bioinformatics and Biology Insights 2022; 16, 11779322221125605.
[49] RK Tiwari, A Brown, N Sadeghiani, AN Shirazi, J Bolton, A Tse, G Verkhivker, K Parang and G Sun. Design, synthesis, and evaluation of dasatinib-amino acid and dasatinib-fatty acid conjugates as protein tyrosine kinase inhibitors. ChemMedChem 2017; 12(1), 86-99.
[50] M Arya, A Tiwari, DB Singh and G Taj. Computational study of lactucine and its derivatives to investigate its anti-cancerous properties targeting apoptosis-inducing protein. Letters in Drug Design & Discovery 2023; 21(7), 1137-1147.
[51] HR Trivedi and PK Puranik. Chlorogenic acid-optimized nanophytovesicles: A novel approach for enhanced permeability and oral bioavailability. Future Journal of Pharmaceutical Sciences 2023; 9, 116.
[52] Y Yan, Kulsoom, Y Sun, Y Li, Z Wang, L Xue and F Wang. Advancing cancer therapy: Nanomaterial-based encapsulation strategies for enhanced delivery and efficacy of curcumin. Materials Today Bio 2025; 33, 101963.
[53] L Xing, ZR Tan, JL Cheng, WH Huang, W Zhang, W Deng, CS Yuan and HH Zhou. Bioavailability and pharmacokinetic comparison of tanshinones between two formulations of Salvia miltiorrhiza in healthy volunteers. Scientific Reports 2017; 7, 4709.