Trends
Sci.
2026;
23(7):
12982
CRISPR/Cas9 Editing of the β-Globin Locus in Sickle Cell Disease and
β-Thalassemia: Mechanistic Rationale, Clinical Evidence, and Future Directions
Thi Anh Ha1, Thi Nga Nguyen1, Tuan Anh Nguyen2, Thu-Thao Thi Huynh1,*,
Anh-Duy Hoang Nguyen3 and Minh Trong Quang3
1Department of Hematology, Faculty of Medical Laboratory, Hong Bang International University,
Ho Chi Minh City, Vietnam
2Molecular Biomedical Center, University Medical Center of Ho Chi Minh City - Branch 2,
Ho Chi Minh City, Vietnam
3School of Pharmacy, University of Medicine and Pharmacy at Ho Chi Minh City, Ho Chi Minh City, Vietnam
(*Corresponding author’s e-mail: [email protected])
Received: 11 December 2025, Revised: 28 January 2026, Accepted: 10 February 2026, Published: 5 March 2026
Abstract
Sickle cell disease (SCD) and β‑thalassemia rank among the world’s most frequent single-gene disorders and remain the leading causes of illness and early death, even with better supportive care. Both conditions stem from harmful mutations in the β‑globin gene (HBB) and are prime candidates for gene-editing therapies, as fetal hemoglobin (HbF) can greatly reduce disease severity. This review first explains the molecular basis of β-hemoglobinopathies and the processes that govern Hb switching, focusing on BCL11A, HBG1/HBG2 promoters, and α-globin as therapeutic targets. This review then examined CRISPR/Cas9 techniques and delivery methods used to edit hematopoietic stem and progenitor cells outside the body, contrasting direct HBB correction with approaches that reactivate HbF and with new multiplex editing strategies. Clinical data from therapies targeting the BCL11A enhancer or the HBG promoter in patients with SCD and transfusion-dependent β‑thalassemia are reviewed, highlighting high rates of transfusion independence, marked reductions in vaso-occlusive crises, and safety primarily limited by conditioning-related toxicity. The article also discusses genomic risks, ethical concerns, cost, and access challenges, and suggests future avenues, such as base and prime editing, in vivo delivery, and safer conditioning regimens. In summary, CRISPR-based editing of the β-globin locus offers a realistic functional cure for selected patients, while underscoring the need for long-term follow-up and fair global implementation.
Keywords: β-thalassemia, β-globin locus, CRISPR/Cas9, fetal hemoglobin, hematopoietic stem cell gene editing, sickle cell disease
Introduction
Sickle cell disease (SCD) and β-thalassemia are the most common inherited disorders that affect β‑globin production and are major causes of sickness and early death worldwide [1–3]. These β-hemoglobinopathies are especially frequent in sub-Saharan Africa, the Middle East, the Indian subcontinent, and the Mediterranean, but migration and improved survival have increased their incidence in affluent nations [1,3,4]. SCD results from a single missense change in the β‑globin gene (HBB), which promotes the polymerization of hemoglobin (Hb) S (HbS), leading to chronic hemolytic anemia, periodic vaso-occlusive attacks, and ongoing damage to multiple organs [2,3]. β‑thalassemia encompasses a variety of conditions in which β‑globin chain synthesis is reduced or absent due to multiple HBB mutations, leading to ineffective red blood cell production, transfusion-dependent anemia, and iron-overload complications [1,4]. Despite advances in newborn screening, infection control, transfusion management, and iron-chelation therapy, many patients still experience shorter lives and significant impairments in quality of life [2,3].
For patients with severe SCD or transfusion‑dependent β-thalassemia, allogeneic hematopoietic stem‑cell transplantation (HSCT) remains the only proven, long‑term cure [2,5]. However, HSCT is limited by a shortage of suitably matched donors and by serious risks, including graft-versus-host disease, transplant-related death, and late sequelae such as infertility and secondary cancers [5]. Recently, the addition of lentiviral-vector-driven genes has offered a powerful alternative, allowing autologous hematopoietic stem and progenitor cells (HSPCs) to produce functional β‑globin or anti‑sickling globin variants [6,7]. Early clinical studies have shown that this strategy can eliminate the need for transfusions in many β-thalassemia patients and markedly reduce vaso-occlusive crises in SCD [6,7]. Gene-addition therapies are complex and expensive, require myeloablative conditioning, and pose theoretical threats of insertional mutagenesis, clonal dominance, and long-term genotoxicity [6]. These challenges have spurred interest in more precise approaches that directly edit patients’ globin genes.
CRISPR/Cas9 has revolutionized the treatment of single-gene disorders. This system uses a programmable RNA-directed endonuclease that creates double‑strand breaks (DSBs) at defined genomic sites, which the cell repairs via natural non-homologous end-joining (NHEJ) or homology-directed repair (HDR) pathways [8]. Compared with earlier tools, such as zinc-finger nucleases and transcription‑activator-like effector nucleases, CRISPR/Cas9 is simpler to design, more scalable, and can simultaneously target many sites [8]. β-hemoglobinopathies are particularly good candidates for CRISPR/Cas9 because disease-causing mutations occur only in blood-forming cells; the relevant stem and progenitor cells can be removed, edited outside the body, and returned, and Hb switching provides multiple logical intervention points [9].
Therapeutic approaches for this condition can be classified into two categories: One strategy aims to repair the primary HBB mutation or otherwise restore normal β‑globin synthesis at the adult locus [9]. The other, conceptually different, leverages the ongoing production of fetal Hb (HbF) as a natural disease modifier, mimicking the hereditary persistence of HbF (HPFH) [9,10]. This is achieved by disrupting regulatory elements, such as the erythroid-specific enhancer of BCL11A, a chief repressor of γ‑globin, or by editing binding motifs in the HBG1 and HBG2 promoters [9–12]. Preclinical studies have shown that both methods can re-establish balanced globin chain production, block HbS polymerization, and rectify ineffective erythropoiesis in model systems [9–11]. For clarity, we use HbF induction and γ-globin reactivation synonymously throughout, as both refer to therapeutic derepression of HBG1/HBG2 expression, thereby increasing HbF production.
Accumulating evidence from these programs led to a landmark regulatory achievement. In late 2023, exagamglogene autotemcel (exa-cel), an ex vivo CRISPR/Cas9 therapy that targets the BCL11A erythroid enhancer, became the first gene-editing product to receive marketing approval for SCD and transfusion-dependent β‑thalassemia, receiving endorsements in multiple regions. These approvals are widely viewed as heralding the start of a new era of somatic genome editing [13,14]. Nevertheless, they also highlight unresolved issues around long-term safety, such as off-target and on-target genomic changes, clonal behavior, and conditioning-related toxicity, together with ongoing challenges in production, cost, and equitable global distribution [8,13,14].
In this context, a comprehensive and critical synthesis of CRISPR/Cas9‑based approaches for SCD and β‑thalassemia is warranted. This review (1) examines the molecular pathology of β-hemoglobinopathies and the biology of Hb switching that underpins current gene-editing strategies, (2) describes the major CRISPR/Cas9 approaches targeting the β-globin locus and HbF regulators, (3) summarizes available clinical trial data for ex vivo-edited HSPC products in SCD and β‑thalassemia, and (4) evaluates key safety, ethical, and implementation issues. By integrating mechanistic insight with emerging clinical experience, this review clarifies the current status of CRISPR/Cas9 for β-hemoglobinopathies. It outlines priorities for developing safer, more effective, and more accessible gene-editing therapies.
Molecular basis of β-hemoglobinopathies and hemoglobin switching
The human β-globin locus, positioned on chromosome 11, comprises a series of genes that are activated in a developmental sequence (ε, Gγ, Aγ, δ, and β) and sits downstream of a locus control region made up of DNase I-hypersensitive sites that interact with each globin promoter in a stage-specific manner to drive high-level expression in erythroid cells (Figure 1(A)) [15]. During embryogenesis, ε‑globin predominates, followed by γ‑globin in fetal life and β‑globin in postnatal erythropoiesis. The temporal switch is achieved by the progressive repositioning of the locus control region from the 5’ embryonic and fetal genes to the adult HBB, accompanied by chromatin remodelling and transcription factor binding [15]. This tightly regulated system ensures balanced production of α- and β-like globin chains, which are essential for Hb assembly and red cell survival. Perturbations in gene structure or regulatory control at the β-globin locus, whether through coding mutations in HBB or changes to regulatory elements, directly produce the quantitative and qualitative defects that characterize SCD and β-thalassemia [15].
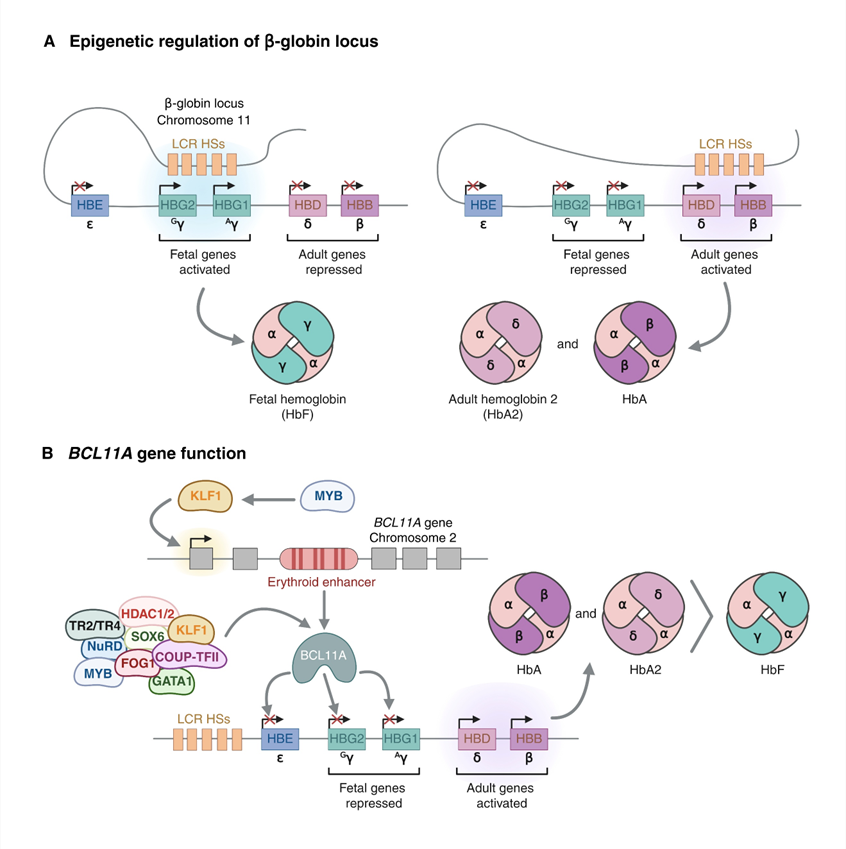
Figure 1 Developmental regulation of the β-globin locus and key regulators of hemoglobin switching. (A) Organization of the human β-globin gene cluster on chromosome 11 and its interaction with the locus control region across embryonic, fetal, and adult life. (B) Structure of the BCL11A locus on chromosome 2, highlighting erythroid-specific enhancers that drive BCL11A expression in red cell precursors and mediate γ-globin repression.
Hb composition follows a predictable developmental pattern: In the womb and at birth, HbF (α₂γ₂) is the main form, but its amount falls rapidly during the first year after birth as adult Hb (HbA, α₂β₂) becomes dominant [15]. By adulthood, HbF usually makes up less than 1% - 2% of total Hb, while HbA accounts for more than 95% [15]. The symptoms of β-hemoglobin disorders mirror this timing: newborns with SCD or β-thalassemia major are often asymptomatic at birth and develop problems only after HbF declines, and the defective or missing adult β-globin takes effect [15,16]. The natural condition of HPFH provides a compelling example of the benefit of continued γ-globin production. People with HPFH maintain high HbF levels into adulthood and usually experience milder SCD or β‑thalassemia, even though they carry disease‑causing HBB mutations [15,17]. These observations support modern therapies that aim to recreate HPFH-like states by reactivating γ-globin transcription or preventing its silencing.
The fetal-to-adult Hb transition is controlled by a network of transcription factors and chromatin regulators that influence the γ- and β-globin promoters and their relationship with the locus control region [15–17]. BCL11A plays a central role in repressing HbF (Figure 1(B)). Initial genomewide association studies linked common variants near BCL11A to differences in HbF levels and the severity of β-hemoglobin disorders among people [17]. Later functional experiments confirmed that BCL11A must be present in erythroid cells to silence γ-globin, and that decreasing BCL11A in a graded fashion can raise HbF levels while leaving overall red cell production largely intact [17]. Liu et al. showed that BCL11A directly binds to specific motifs duplicated in the HBG1 and HBG2 promoters, and that binding to a distant motif essential for HPFH keeps those promoters turned off [18]. When this motif was altered in human erythroid cells, it disrupted BCL11A binding and allowed γ‑globin to be expressed, establishing a clear molecular connection between BCL11A, the HBG promoters, and the Hb switch [18]. Concurrently, researchers identified erythroid-specific enhancers inside the BCL11A locus that steer its expression in red cell precursors, providing a tissue-specific target for gene editing that preserves BCL11A function in other cell types [17]. Other factors, such as KLF1, MYB, ZBTB7A (LRF), and polycomb and NuRD complex components, work with BCL11A as part of a larger epigenetic network that shapes the chromatin structure at the β-globin locus [15,16]. Recent studies have broadened this network, uncovering additional HbF modulators through CRISPR-based functional screening and large-scale genomic analyses [16]. Taken together, these findings offer multiple rational therapeutic targets and explain why altering BCL11A or its binding sites has emerged as the leading strategy for CRISPR/Cas9-driven HbF induction.
In SCD, a single Glu6Val substitution in β‑globin produces HbS, which polymerizes upon deoxygenation, bending red blood cells into the classic sickle shape, stiffening their membrane, and triggering repeated cycles of deformation and rehydration that shorten the red blood cell lifespan (Figure 2(A)) [2]. Consequently, chronic intravascular and extravascular hemolysis leads to anemia, releases free Hb and heme, scavenges nitric oxide, and triggers endothelial dysfunction. Adhesive and rigid cells also promote microvascular occlusion, ischemia-reperfusion injury, and a persistent inflammatory state [2]. HbF strongly mitigates this sequence by interfering with HbS polymerization when co-assembled in the same tetramer or in the same erythrocyte. Even modest elevations in HbF, particularly when distributed pancellular across most erythrocytes, markedly diminish polymerization, sickling, and vaso-occlusive events [2,15,16]. These observations explain why individuals with different HbF levels show a wide range of clinical severity and underpin the therapeutic rationale for drugs and gene-editing strategies that aim to raise HbF rather than directly modify the sickle mutation.
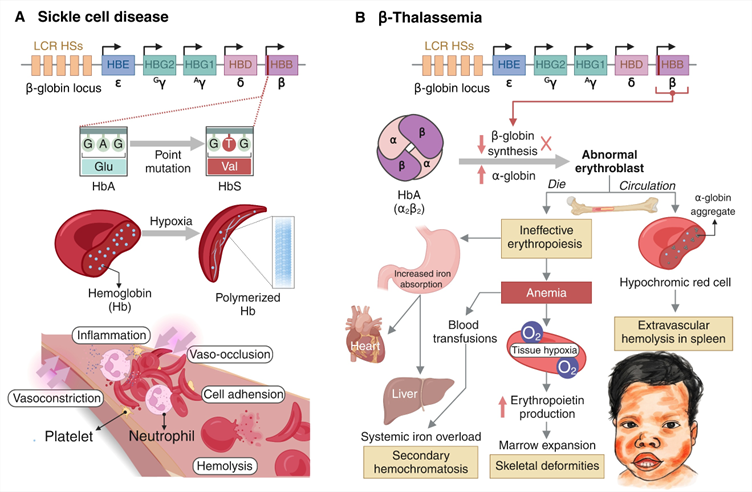
Figure 2 Pathophysiological mechanisms of sickle cell disease and β-thalassemia. (A) Glu6Val mutation in the β‑globin gene results in hemoglobin S polymerization, red cell sickling, hemolysis, vaso-occlusion, and progressive multiorgan damage. (B) Imbalance between α- and β-like globin chains in β-thalassemia, resulting in ineffective erythropoiesis, marrow expansion, extramedullary hematopoiesis, and hepcidin suppression with secondary iron overload.
β-thalassemia arises from mutations that either reduce (β⁺) or eliminate (β⁰) β‑globin synthesis, leading to an excess of α-globin chains relative to β-like chains (Figure 2(B)) [1,4]. On the cellular level, the unpaired α-globin aggregates in developing erythroblasts, generating reactive oxygen species, damaging cellular membranes, and triggering apoptosis [19,20]. These events account for the hallmark of β-thalassemia, ineffective erythropoiesis, characterized by a surge of immature erythroid precursors that die prematurely within the marrow and fail to produce enough mature red cells [19,20]. Ineffective erythropoiesis produces systemic consequences, including anemia and tissue hypoxia, which stimulate erythropoietin release and expand the erythroid compartment, whereas erythroid regulators, such as erythroferrone, suppress hepatic hepcidin, resulting in heightened intestinal iron absorption and increased iron release from macrophages [19,20]. The combination of transfusion-dependent iron loading and hepcidin suppression leads to progressive parenchymal iron overload, which contributes to endocrine, hepatic, and cardiac complications [4,19,20]. This cascade shows that β-thalassemia is not merely an anemic disorder but a disease of disrupted erythropoiesis and iron metabolism. From a gene-editing perspective, restoring balanced β-globin production or achieving sufficiently high γ-globin expression could correct the chain imbalance, reduce ineffective erythropoiesis, and re-establish normal iron homeostasis.
In summary, β‑thalassemia and SCD are caused by the abnormal expression or structure of β-like globin chains, with HbF acting as an important disease modifier. The precise developmental regulation of the β-globin locus, together with the critical roles of BCL11A and related repressors in silencing γ-globin, delineates a highly tractable set of genomic targets for CRISPR/Cas9-based therapies.
CRISPR/Cas9 technology and delivery strategies for hematopoietic stem cell editing
The CRISPR/Cas9 system used in mammalian cells is derived from type II bacterial adaptive immune systems. It consists of a programmable guide RNA (gRNA) that directs the Cas9 endonuclease to a complementary genomic sequence adjacent to a protospacer adjacent motif (PAM) [8,21]. Streptococcus pyogenes Cas9 (SpCas9) employs its HNH and RuvC nuclease domains to introduce a site-specific DSB when bound [21]. The break is repaired by endogenous DNA repair mechanisms, mainly NHEJ or microhomology-mediated end joining (MMEJ), which often generate small insertions or deletions (indels) at the cut site, or by HDR if a donor template is supplied (Figure 3(A)) [22]. In treating β‑hemoglobinopathies, indels created via NHEJ/MMEJ are intentionally used to inactivate regulatory elements, such as the BCL11A erythroid enhancer or HBG1/2 promoter binding motifs, thereby lifting repression of γ‑globin expression [9,10,18]. However, HDR has the potential to correct the sickle mutation or repair β‑thalassemia alleles at the HBB locus, although its efficiency in quiescent hematopoietic stem cells (HSCs) remains relatively low [22,23]. Strategies to enhance HDR in long-term repopulating HSCs include refined template design, cell-cycle synchronization, and modulation of DNA repair pathways [22,23].
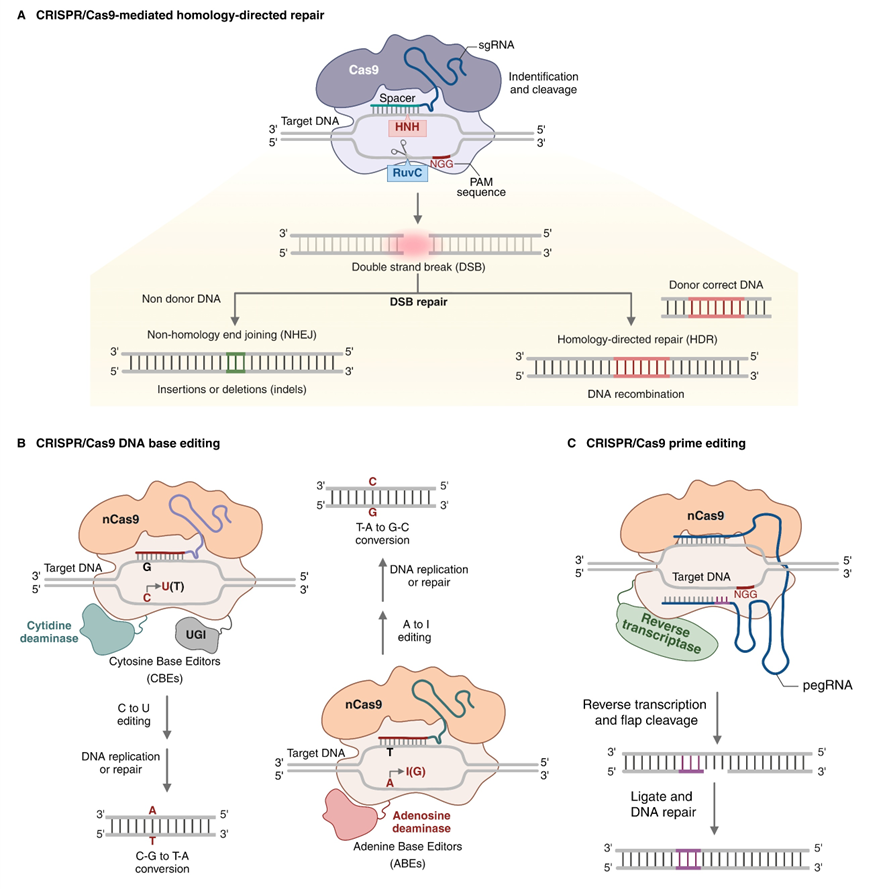
Figure 3 Genome-editing platforms targeting the β-globin locus. (A) Conventional CRISPR/Cas9 nuclease editing: Cas9-guide RNA-induced double-strand breaks repaired by non-homologous end-joining/microhomology-mediated end joining to disrupt regulatory elements. (B) Cytosine and adenine base editors that couple a Cas9 nickase to DNA deaminases, enabling targeted C→T or A→G conversions without generating double-strand breaks. (C) Prime editors that fuse a Cas9 nickase to a reverse transcriptase and use a prime-editing guide RNA to install defined substitutions, small insertions, or deletions without exogenous donor DNA. Clinically, these platform choices determine the balance between editing efficiency and long-term genotoxic risk in hematopoietic stem and progenitor cells, shaping which approaches are most suitable for near-term translation versus next-generation therapies.
DSB-based editing is linked to a range of on‑target and off‑target outcomes. Indels can generate frameshift mutations or in-frame variants that may retain partial function at the intended locus [21,22]. Large deletions, inversions, or complex re-arrangements have also been observed at on‑target sites, raising concerns about genotoxicity when editing occurs in long‑lived stem cells [24]. Off‑target cleavage, which is driven by partial complementarity between gRNA and unintended genomic sites, can similarly produce disruptive mutations or structural variants [21,24]. Strategies to improve specificity include designing high-fidelity Cas9 variants with reduced off-target activity, using truncated or chemically modified gRNAs, and unbiased genome-wide profiling followed by computational or empirical prediction of off-target sites [21,24,25]. From a therapeutic standpoint, ex vivo editing of HSPCs permits rigorous quality control of the edited product, including on-target editing efficiency assessment, off-target profiles at predicted sites, and vector integration before reinfusion into patients [10,24]. Nonetheless, the possibility of rare, undetected events that could contribute to clonal expansion or malignant transformation underscores the need for long‑term surveillance in treated individuals [24,25].
Several molecular formats of CRISPR/Cas9 have been used for ex vivo editing of HSPCs, each with its own advantages and disadvantages: Early studies used plasmid DNA encoding Cas9 and gRNA, which provided sustained enzyme expression but also caused random genomic integration and prolonged nuclease activity [22]. This approach has largely been replaced by transient delivery of Cas9 mRNA and gRNA or, in most current clinical protocols, by ribonucleoprotein (RNP) complexes made from recombinant Cas9 protein pre-assembled with synthetic gRNA [9,10,22]. RNP delivery offers several therapeutic benefits, including rapid nuclear entry, a high peak of activity, and a brief intracellular half-life, all of which reduce off-target exposure and lower the likelihood of an immune response to Cas9 [22,26]. The electroporation of Cas9-gRNA RNPs into mobilized CD34⁺ HSPCs has become the predominant strategy in trials targeting the BCL11A enhancer, achieving editing efficiencies above 70% - 80% at the bulk cell level while preserving engraftment capacity [10,26]. When HDR is required, a donor template is typically co-delivered via single-stranded oligodeoxynucleotides or recombinant adeno-associated virus serotype 6 (AAV6), which efficiently transduces HSPCs and supplies high-copy donor DNA without genomic integration [22,23,27]. Therefore, optimizing ex vivo editing demands a balance between efficiency and stem-cell preservation, with key variables including the duration and composition of pre-stimulation culture, cytokine mixtures, electroporation settings, and postediting recovery periods [23,26]. Excessive culture or strong proliferative cues can drive differentiation and impair long-term engraftment, whereas insufficient activation may limit editing within the most primitive HSC compartment [23,26].
Stable, long-term editing of HSCs that can repopulate is crucial for ongoing clinical benefit. Xenograft experiments have shown that bulk CD34⁺ cell editing rates often overstate the true editing level in the stem cell fraction, which drives long‑term hematopoiesis [23,26]. Introducing DSBs in these cells can provoke p53-mediated DNA damage responses, leading to a brief cell cycle pause, apoptosis, or selective expansion of rare p53-deficient clones [24,28,29]. Such selection pressures could, in theory, allow clonal dominance and cancer, echoing earlier worries about insertional oncogenesis from retroviral gene therapy [24,28]. Clinical protocols typically limit nuclease exposure to a short window using RNP delivery, sharpen gRNA precision, and exclude cells that show clear cytogenetic defects or major DNA damage from the final product to reduce these risks [10,24]. Ongoing studies have explored temporary p53 suppression, chromatin-focused methods, and non-cleaving editors to maintain HSC fitness while delivering therapeutically significant editing [24,28].
Recognition of the genotoxic potential of DSBs has driven the development of CRISPR-derived editors that modify DNA without generating frank DSBs. Base editors, which fuse catalytically impaired or nickase Cas9 to cytidine or adenosine deaminases, enable precise C→T or A→G conversions within a defined editing window (Figure 3(B)) [30]. Prime editors combine a Cas9 nickase with a reverse transcriptase and a prime-editing gRNA to install a broader range of substitutions, small insertions, and deletions without requiring exogenous donor templates (Figure 3(C)) [30]. Although these technologies are at an earlier stage of development than conventional Cas9 nucleases, they are highly relevant to β-hemoglobinopathies. Base or prime editors could correct sickle mutations or recreate HPFH-associated point mutations in HBG promoters while reducing the risk of large deletions and translocations [30,31]. Proof-of-concept studies in human HSPCs have demonstrated efficient base editing at β-globin loci with preservation of engraftment in xenograft models. However, clinical translation has not yet kept pace with nuclease-based platforms [31]. As these tools mature, they may eventually complement or supplant DSB-based CRISPR/Cas9 approaches for β-hemoglobinopathies, especially if they can be integrated with improved delivery systems and safer conditioning regimens.
Taken together, the current genome-editing tools provide a flexible set of options for altering the β‑globin locus and its regulators in autologous HSPCs. However, the unavoidable trade-offs among editing efficiency, precision, genotoxicity, and preservation of HSC function continue to guide the design of clinical protocols and drive the development of next-generation editors.
Therapeutic CRISPR/Cas9 strategies in sickle cell disease and β-thalassemia
An intuitive approach to treating β‑hemoglobinopathies is to repair the core HBB mutation or otherwise restore normal β‑globin expression at the adult locus. Accurate HBB editing would enable normal HbA production within the cell’s own regulatory setting, eliminating abnormal expression and preserving the typical developmental pattern of the β‑globin locus (Figure 4(A)) [9,22]. Most HBB-correction methods use CRISPR/Cas9 to create a DSB near the disease-causing change and provide an HDR external donor template [22,23]. Early studies addressed the sickle mutation in human CD34⁺ cells, combining CRISPR/Cas9 with AAV6 donor templates to achieve efficient gene correction and restore adult β‑globin levels in erythroid progeny and xenograft models [22]. Hoban et al. reported a similar correction of the sickle mutation in patient-derived HSPCs, where edited cells produced normal Hb and showed improved hematologic parameters after transplantation into immunodeficient mice [23]. More recently, Cosenza et al. successfully corrected the common β⁰³⁹‑thalassemia nonsense mutation using CRISPR/Cas9 in erythroid progenitors and HSPCs, re‑establishing β‑globin production and normalizing globin-chain ratios in vitro [32]. Although these encouraging results are promising, several challenges are slowing the rapid clinical rollout of HDR-based HBB gene edits. First, although HDR works best in actively dividing cells, long-term repopulating HSCs remain largely quiescent, yielding lower editing rates in the stem-cell niche than in bulk CD34⁺ populations [22,23]. Second, β‑thalassemia arises from various HBB mutations; thus, tailoring donor DNA to each mutation could be necessary, complicating product standardization [4,9]. Third, DSBs in HBB can cause sizeable deletions or structural changes in this highly transcribed gene, raising concerns about on-target genotoxicity [24,33]. Direct HBB correction remains in the preclinical phase, and current clinical efforts focus on mutation-agnostic strategies that reactivate HbF.
The most advanced CRISPR/Cas9 approach for β-hemoglobinopathies targets the erythroid-specific enhancer of BCL11A, a key transcriptional repressor of γ‑globin [17]. Genome-wide association studies first linked common variants at this enhancer to inter-individual differences in HbF levels, and functional work confirmed that the region is essential for BCL11A expression in erythroid cells but is dispensable in other lineages (Figure 4(B)) [17,34]. Canver et al. used Cas9-mediated saturating mutagenesis to dissect the BCL11A enhancer and found a critical motif-containing short core region [33]. Disruption of these motifs markedly reduces BCL11A expression and derepresses γ‑globin in human erythroid cells. Subsequent studies in primary CD34⁺ HSPCs showed that CRISPR/Cas9 enhancer editing induces robust HbF expression while preserving multilineage differentiation, supporting its suitability as a therapeutic target [34,35]. Antoniani et al. used CRISPR/Cas9 to disrupt the BCL11A erythroid enhancer in human β-thalassemia erythroid progenitors, achieving substantial γ‑globin induction and correcting the imbalance in globin chain expression [36]. Psatha et al. extended these findings by editing the enhancer in HSPCs from patients with β‑thalassemia major, demonstrating in vitro reactivation of HbF and amelioration of the disease phenotype [37]. Mouse models carrying human β‑globin loci have confirmed that disruption of the HSC enhancer leads to sustained HbF expression in circulating erythrocytes and corrects anemia [33]. These preclinical data formed the basis for ex vivo clinical protocols in which mobilized autologous CD34⁺ cells are edited with Cas9-gRNA RNPs targeting the BCL11A enhancer, followed by myeloablative conditioning and reinfusion [10,11]. In a pivotal phase 1 - 2 trial, Frangoul et al. reported that patients with transfusion-dependent β‑thalassemia or severe SCD treated with BCL11A enhancer-edited HSPCs achieved high levels of HbF, increased total Hb, transfusion independence in almost all β‑thalassemia participants, and resolution of severe vaso-occlusive crises in SCD [10]. Fu et al. showed that the same strategy is feasible and effective in pediatric patients with β⁰/β⁰ β-thalassemia, with edited HSPCs durably engrafting and sustaining high HbF levels [11]. The appeal of BCL11A enhancer editing lies in several features: it is mutation-independent, leverages human genetic evidence for safety and efficacy, and restricts BCL11A downregulation to erythroid cells, thereby preserving its functions in B cells and other tissues [17,34]. Potential limitations include variable editing patterns across alleles, uncertainty about the long-term consequences of BCL11A modulation, and continued reliance on intensive conditioning and ex vivo manufacturing. Nevertheless, BCL11A enhancer editing has the most extensive clinical track record and serves as the basis for the first approved CRISPR/Cas9-based therapy among current approaches.
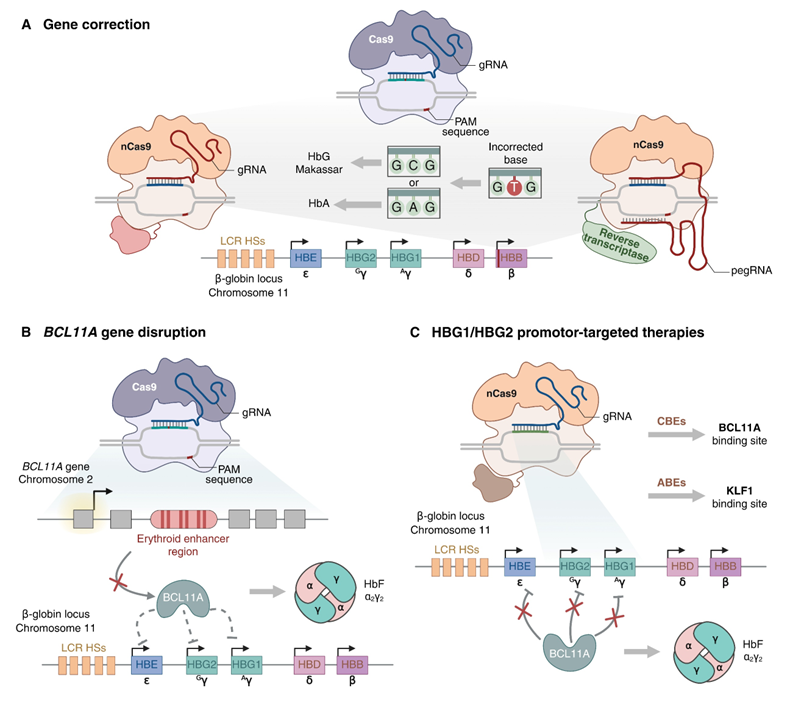
Figure 4 CRISPR-based therapeutic strategies for β-hemoglobinopathies. (A) Homology-directed repair-mediated correction of pathogenic β‑globin gene variants to restore physiological adult hemoglobin A synthesis. (B) Disruption of the erythroid-specific BCL11A enhancer to downregulate BCL11A in erythroid cells, derepress γ-globin, and increase fetal hemoglobin. (C) Editing of HBG1/HBG2 promoters to mimic hereditary persistence of fetal hemoglobin alleles and reactivate fetal hemoglobin, with multiplex approaches incorporating α-globin modulation to rebalance globin chain synthesis in β-thalassemia. Clinically, these strategies map onto current therapeutic products, with fetal hemoglobin-induction approaches (BCL11A enhancer or HBG promoter editing) representing the most mature routes to functional cure in sickle cell disease and transfusion-dependent β-thalassemia.
A complementary strategy targeting the promoters of the γ‑globin HBG1 and HBG2 genes. Natural HPFH mutations in these promoters destroy binding sites for BCL11A and other repressors and are associated with high HbF levels and milder disease in people who also inherit β‑hemoglobinopathies (Figure 4(C)) [15,17,38]. This experiment suggests that mimicking HPFH-associated promoter variants could safely increase HbF levels. CRISPR/Cas9 has validated this idea in several systems: Weber et al. introduced HPFH-like mutations into the distal γ‑globin promoter via nuclease editing, restoring HbF expression in adult‑type erythroid cells and improving disease markers in SCD models [38]; Traxler and colleagues targeted a 13-nucleotide segment upstream of the HBG promoters, and showed that deletion or inversion of this region provokes robust γ‑globin reactivation along with altered chromatin contacts at the β‑globin locus [39]. More recent work has employed base editing to scan the HBG promoters for point mutations that elevate HbF, uncovering novel HPFH‑like variants that could serve as future therapeutic templates [5,33]. Lu et al. demonstrated that the precise insertion of natural HPFH mutations into HBG promoters in human HSPCs using CRISPR/Cas9 and AAV6‑mediated HDR induces strong γ‑globin expression while preserving engraftment in xenograft models [40]. Translation into the clinic has begun with OTQ923, an autologous CD34⁺ HSPC product edited using CRISPR/Cas9 to disrupt the HBG1 and HBG2 promoters [41]. Sharma et al. reported that three treated participants showed stable engraftment, sustained HbF increases (about 19% - 27% of total Hb), a broad distribution of HbF across red cells, and clinical improvement over 6 - 18 months of follow-up in an early phase study of adults with severe SCD [41]. No off‑target mutations were detected at predefined candidate loci, although the small cohort and limited follow‑up preclude definitive long-term safety conclusions. HBG promoter targeting offers theoretical advantages relative to BCL11A enhancer editing: It acts closer to the effector genes, directly emulates benign HPFH alleles, and avoids disrupting upstream regulators, such as BCL11A, which play wider roles in hematopoiesis and immunity [17,38]. However, promoter editing may produce a more complex spectrum of allelic outcomes, such as large deletions affecting both HBG1 and HBG2, whose long-term impacts on locus architecture and regulation remain undefined, and clinical experience with HBG-targeted products is still more limited than with BCL11A-focused approaches [24,39].
In β‑thalassemia, the severity of the disease is determined not only by the total absence of β‑globin but also by how much the α‑ and β‑like chains are out of balance [19]. This has led to increased interest in therapies that lower α‑globin levels, thereby restoring a healthier chain synthesis balance even when β‑globin cannot be fully restored [19,26]. The fact that co‑inherited α‑thalassemia improves β‑thalassemia symptoms provides a clinical rationale for such strategies [19]. A notable study used CRISPR/Cas9 to delete the main α‑globin enhancer (MCSR2) in patient-derived primary CD34⁺ cells, effectively mimicking a natural α‑thalassemia mutation and reducing α‑globin expression [33]. These edited erythroid cells achieved a better globin chain balance and exhibited lower oxidative-damage markers, indicating that targeting α‑globin alone can address key features of β‑thalassemia [33]. Building on this idea, Pavani et al. performed multiplex editing of the α‑globin locus in HSPCs taken from β-thalassemia patients, again using CRISPR/Cas9 [34]. Their strategy combined the deletion of HBA2 to lower α-globin with targeted insertion of a β‑globin transgene driven by the HBA2 promoter, thereby correcting both the β‑globin deficiency and the chain imbalance simultaneously [35]. In both in vitro and in vivo studies, this approach restored Hb production and improved erythroid parameters, supporting the feasibility of multiplex editing as a therapeutic option, albeit technically demanding [23]. These α‑globin‑targeted approaches highlight the flexibility of genome editing in β‑thalassemia, showing that diminishing excess α‑globin can be as critical as boosting β‑like chains. They also suggest the promise of truly patient-specific multiplex editing, combining HBB correction, HbF reactivation, and α‑globin silencing, while acknowledging that each added edit increases both complexity and risk [21,23,30].
Overall, three main CRISPR/Cas9 approaches have been identified for treating β‑hemoglobinopathies: (i) direct correction of HBB mutations, (ii) reactivation of HbF by editing the BCL11A enhancer, and (iii) reactivation of HbF by editing the HBG promoter, with α-globin modulation added mainly for β-thalassemia. Directly correcting HBB is conceptually attractive because it restores normal adult Hb levels; however, it is constrained by low HDR efficiency, disease-causing allele heterogeneity, and the potential for on-target genotoxicity [22,23,27,42]. The BCL11A enhancer is mutation-agnostic, supported by strong human genetic and functional studies, and has been validated in multicenter clinical trials. However, it disrupts an upstream regulator that acts outside the β‑globin locus and still requires intensive conditioning and ex vivo cell processing [10,11,17,34]. Editing the HBG promoter more closely mimics natural HPFH mutations and keeps BCL11A intact in non-erythroid tissues; however, clinical data are still lacking, and the full spectrum of structural changes and long-term safety remains unknown [38,39,41]. At present, HbF-induction strategies, particularly those targeting the BCL11A enhancer, represent the most advanced clinical application of CRISPR/Cas9 in SCD and β‑thalassemia. Nevertheless, advances in base and prime editing, deeper insights into globin gene regulation, and experience with α-globin targeting suggest that the therapeutic toolkit will likely expand further and may eventually provide personalized combinations of edits tailored to disease genotype, severity, and comorbidities [9,24,30–32].
To facilitate direct comparison across these approaches, Table 1 summarizes the principal CRISPR/Cas9-based strategies for β-hemoglobinopathies, highlighting their genomic targets, mechanisms of action, clinical development stage, key advantages, and major limitations. This comparative framework underscores how different editing strategies address different aspects of disease biology and clarifies the trade-offs that currently shape clinical decision-making.
Table 1 A comparative overview of major CRISPR-based therapeutic strategies for β-hemoglobinopathies.
Strategy |
Primary genomic target |
Mechanism of therapeutic benefit |
Clinical maturity |
Key advantages |
Principal limitations |
Direct HBB correction |
The HBB coding region |
HDR-mediated repair of pathogenic β-globin mutations restores HbA synthesis |
Preclinical/early translational |
Physiologic restoration of HbA; preservation of native globin regulation |
Low HDR efficiency in long-term HSCs; mutation-specific design; on-target rearrangement risk |
BCL11A enhancer disruption |
Erythroid-specific enhancer of BCL11A |
NHEJ-mediated BCL11A loss repression induces robust HbF expression |
Clinical late-stage; regulatory approval |
Mutation-agnostic; strong human genetic validation; high HbF induction |
Disrupts upstream regulator; relies on myeloablation and ex vivo processing |
HBG1/HBG2 promoter editing |
γ-globin promoters |
Mimics HPFH mutations by disrupting repressor binding sites and reactivating HbF |
Early clinical |
Directly emulates benign human variants and preserves BCL11A function elsewhere |
More heterogeneous editing outcomes and limited long-term clinical data |
α-globin modulation |
α-globin enhancers or genes |
Reduces α/β chain imbalance, improving erythropoiesis in β-thalassemia |
Preclinical |
Core pathophysiology of β-thalassemia; mutation-independent |
Risk of inducing clinically significant α-thalassemia; added complexity |
Multiplex editing strategies |
Multiple loci (e.g., HBB + α-globin or HbF regulators) |
Combination of correction, HbF induction, and chain rebalancing |
Preclinical |
Highly customizable; potential for superior phenotypic correction |
Increased technical complexity and cumulative genotoxic risk |
Base/prime editing |
HBB or other regulatory elements |
Precise nucleotide changes without DSBs |
Preclinical |
Risk reduction of large deletions and translocations; high precision |
Delivery challenges, off-target base edits, and limited clinical experience |
Abbreviations: DSB, double-strand break; HBB, β-globin gene; HbF, fetal hemoglobin; HbA, adult hemoglobin A; HSCs, hematopoietic stem cells; HSPCs, hematopoietic stem and progenitor cells; HDR, homology-directed repair; NHEJ, non-homologous end joining; HPFH, hereditary persistence of fetal hemoglobin.
Clinical translation of CRISPR/Cas9 therapies in sickle cell disease and β-thalassemia
For clarity, after the first mention, CRISPR/Cas9-based products are referred to by their generic names, with development codes or marketed names specified only at the initial introduction. Most CRISPR/Cas9 studies that aim to treat β‑hemoglobinopathies follow a similar ex vivo workflow: Autologous CD34⁺ HSPCs are first mobilized, then collected, edited with Cas9 RNP complexes that target either the BCL11A erythroid enhancer or the HBG1/HBG2 promoters, cryopreserved, and finally reinfused after myeloablative busulfan conditioning [9,10,16,34,41]. In phase 1 - 2 trials, the main focus is on safety and feasibility to achieve successful neutrophil and platelet engraftment. The key efficacy endpoints are changes in total Hb, HbF percentage, HbF-positive red cell distribution, transfusion independence in β‑thalassemia, and a reduction or elimination of vaso-occlusive crises in SCD [10,16,34,41,43]. Early follow-up consistently showed rapid neutrophil and platelet recovery, sustained engraftment of edited cells, and marked increases in HbF [10,11,16,34,41,43]. Nonetheless, the sample sizes are still small, and follow-up is limited compared to the expected lifespan of edited HSCs, so long-term safety and durability should be interpreted with caution [43–47].
The leading CRISPR/Cas9 therapy for β‑hemoglobin disorders is exagamglogene autotemcel (exa-cel; marketed as Casgevy), an autologous CD34⁺ HSC product edited at the erythroid-specific BCL11A enhancer [16,34,48]. In the initial report, two adults, one with transfusion‑dependent β‑thalassemia and another with severe SCD, received exagamglogene autotemcel (exa-cel) and achieved extensive BCL11A enhancer editing, HbF reactivation, and clinically significant improvements, including becoming transfusion-independent and eliminating severe vaso-occlusive crises during early follow-up [10,14]. Subsequent pivotal phase 3 studies, CLIMB THAL‑111 (transfusion‑dependent β‑thalassemia) and CLIMB SCD-121 (severe SCD), broadened the assessment to larger patient groups. In the β-thalassemia trial, 52 participants were treated with exagamglogene autotemcel (exa-cel); an interim analysis showed that 91% of those with adequate follow-up attained durable transfusion independence, usually within weeks of the infusion [48,49]. In the parallel SCD study, exagamglogene autotemcel (exa‑cel) dramatically lowered acute events: among participants followed for at least 12 months, 97% experienced no severe vaso-occlusive crises, while total Hb increased to levels consistent with transfusion independence and improved functional status [16,48,49]. High HbF levels and a broad distribution of HbF-positive red cells accompanied these outcomes [16,49]. Based on these findings, exagamglogene autotemcel (exa-cel) was approved for use in SCD and transfusion‑dependent β‑thalassemia in multiple regions, marking the first licensed CRISPR/Cas9-edited cell therapy [12,13,46]. Short-term to mid-term safety data have mainly reflected expected adverse effects of busulfan conditioning and autologous transplantation, such as febrile neutropenia, mucositis, and infections, with no clear evidence of off-target editing toxicity or treatment-related hematologic cancers to date [10,16,48–50].
Nonetheless, regulators and investigators underscore the need for long-term monitoring because edited HSCs persist throughout life and rare late adverse events may occur [46,47,50]. In addition to exagamglogene autotemcel (exa-cel), several regional initiatives target the BCL11A enhancer. ET-01 is a similar autologous CD34⁺ product developed in China that targets the erythroid BCL11A enhancer using Cas9-mediated genome editing [20]. Preclinical studies showed efficient editing and HbF induction in HSPCs, and early clinical reports in transfusion‑dependent β‑thalassemia are reported in abstracts. Yet, detailed peer-reviewed outcome data remain scarce [20,43]. BRL-101 is another autologous CD34⁺ cell therapy in which the erythroid-specific BCL11A enhancer is disrupted using CRISPR/Cas9. In an open-label study, six children, four of whom carried the β⁰/β⁰ genotype, received BRL-101 after busulfan conditioning. All achieved rapid neutrophil and platelet engraftment, sustained editing in peripheral blood cells, and transfusion independence, with total Hb approaching physiologic levels and HbF exceeding 70% in many cases [11,33]. The safety profile of this study aligned with that of autologous transplantation; serious adverse events were predominantly infectious or cytopenic and resolved with standard management, and no product-related deaths or malignancies occurred during follow-up [11,33]. Updated data continue to confirm the durability of transfusion independence in this pediatric cohort [1,33,45]. Taken together, these studies demonstrate that disruption of the BCL11A enhancer reliably induces high HbF, corrects anemia in β‑thalassemia, and prevents vaso-occlusive events in SCD across diverse ages and genotypes, with a safety profile largely driven by conditioning rather than the editing itself in the short- to intermediate-term [10,11,16,33,46,47,50].
A second group of studies targets the γ‑globin promoters directly to mimic HPFH alleles. OTQ923 is an autologous CD34⁺ cell product in which Cas9 RNPs introduce insertions or deletions at specific sites in both HBG1 and HBG2 promoters, generating a spectrum of allelic variants, including a common approximately 5‑kb deletion that fuses the HBG2 promoter to the HBG1 coding sequence [39,41]. In a phase 1 - 2 trial, three adults with severe SCD received OTQ923 after busulfan conditioning and were followed up for 6 - 18 months [41]. All patients engrafted and maintained HbF reactivation, with HbF comprising about 20% - 25% of total Hb and HbF-positive red cells making up more than 70% of the erythrocyte population [41]. The clinical manifestations of SCD were markedly reduced during the follow-up, and no new safety issues arose beyond those expected from myeloablation and autologous transplantation [41]. Although the cohort was small, HBG promoter editing can produce HbF levels that closely mirror those seen in human HPFH. RM-001 is an autologous CRISPR/Cas9-edited CD34⁺ cell therapy targeting BCL11A-binding sites within the HBG1/HBG2 promoters. It is a related therapy for transfusion‑dependent β‑thalassemia that also uses CRISPR/Cas9 to delete the BCL11A binding site in the HBG1/HBG2 promoters [39,43,44]. The first human report described two β⁰/β⁰ patients who received RM-001 after busulfan conditioning and became transfusion-independent within 1 - 2 months, with higher total Hb and increased HbF levels [43,44]. Later abstracts have expanded the data to at least seven patients, demonstrating consistent engraftment, lasting transfusion independence, and a safety profile mainly driven by conditioning-related events [43–45]. Compared with BCL11A enhancer editing, the clinical evidence for HBG promoter-targeted therapies is limited, with fewer patients and shorter follow-up. Nevertheless, existing data show that promoter editing can trigger extensive HbF production and deliver meaningful hematologic and clinical improvements in both SCD and β‑thalassemia, justifying ongoing development [39,41,43–45].
Taken together, ex vivo CRISPR/Cas9 interventions for β‑hemoglobinopathies exhibit several key outcome patterns. First, almost all β‑thalassemia patients treated in published BCL11A enhancer and HBG promoter studies who have reached adequate follow-up have become transfusion-independent, with total Hb generally stabilizing above 10 g/dL and frequently within the normal range [10,11,33,43–45,48,49]. Second, patients with SCD enrolled in both BCL11A and HBG-targeted programs experience substantial and sustained reductions in vaso-occlusive crises, often eliminating severe events for at least 12 months of follow-up [10,16,41,46,49]. Third, HbF induction is robust across platforms, although the magnitude and distribution differ: BCL11A enhancer editing typically produces HbF levels exceeding 40% with a high proportion of F-cells, whereas HBG promoter editing in OTQ923 yields somewhat lower HbF percentages (approximately one-quarter of total Hb) yet shows a broad F-cell distribution [10,11,16,34,41,43]. Direct head-to-head comparisons between BCL11A enhancer and HBG promoter editing are not yet available, and cross-trial comparisons must be approached cautiously due to variations in patient selection, baseline disease severity, conditioning regimens, and endpoints [41,46,47]. Current evidence indicates that both strategies can achieve a functional cure in a substantial proportion of patients, at least over several years of follow-up [10,11,16,33,41,43–47]. Determining whether one approach offers superior long-term durability, safety, or quality-of-life outcomes will require longer follow-up and larger cohorts.
The most frequent acute toxicities stem from busulfan-induced myeloablation and mirror those seen with conventional autologous transplantation, such as febrile neutropenia, mucositis, transient liver enzyme elevations, infections, and cytopenia-related complications [10,11,16,33,41,43,48,49]. No study has yet documented a definitive case of insertional oncogenesis, editing-driven clonal hematopoiesis, or therapy-related myeloid neoplasms directly linked to CRISPR/Cas9, although follow-up periods remain shorter than the lifespan of hematopoiesis [24,43–45,47]. Targeted deep sequencing and, in some instances, broader assays have been used to assess on- and off-target genomic alterations, generally showing high editing efficiency at the intended sites and low rates of predicted off-target events, thanks to carefully selected gRNAs and transient RNP delivery [10,16,34,41,43]. However, emerging data from model systems reveal large deletions, complex re-arrangements, and chromothripsis after Cas9-induced DSBs, indicating a nonzero theoretical risk that current assays may not fully capture, especially for rare clones that expand slowly over time [24,43–45,47]. Early systematic reviews of CRISPR/Cas9 trials in transfusion‑dependent β‑thalassemia and SCD have shown impressive efficacy; however, small patient numbers and limited follow-up have prevented firm conclusions about long-term safety [43,44,46,47]. Moreover, all existing ex vivo programs use high-dose busulfan, which itself poses risks of infertility, gonadal failure, and secondary cancers, independent of gene editing [16,46,48]. These concerns have driven interest in less toxic conditioning regimens and non-DSB editors, such as base and prime editors, that may reduce genotoxic risk [24,30,31,43,47].
While individual clinical programs are discussed above, direct comparison across studies is challenging due to differences in editing targets, conditioning regimens, patient populations, and follow-up duration. To facilitate synthesis and improve clarity, Table 2 summarizes the major CRISPR/Cas9-based clinical programs for SCD and transfusion-dependent β-thalassemia, comparing their molecular targets, editing strategies, conditioning approaches, patient numbers, follow-up, key efficacy outcomes, and principal safety findings.
Table 2 A comparative summary of major CRISPR/Cas9 clinical programs for β-hemoglobinopathies.
Therapy/Program |
Primary target |
Editing strategy |
Conditioning regimen |
Indication |
No. of patients (reported) |
Follow-up (approx.) |
Key efficacy outcomes |
Exagamglogene autotemcel (exa-cel) |
BCL11A erythroid enhancer |
Cas9 RNP and NHEJ-mediated disruption |
Myeloablative busulfan |
SCD; TDT |
> 100 (combined trials) |
up to ~ 4 years |
Transfusion independence in ~ 90% of patients with TDT; elimination of severe VOCs in ~ 95% of patients with SCD |
BRL-101 |
BCL11A erythroid enhancer |
Cas9-mediated enhancer disruption |
Myeloablative busulfan |
Pediatric TDT |
6 (reported) |
~ 2 years |
Sustained transfusion independence; HbF often > 70% |
ET-01 |
BCL11A erythroid enhancer |
Cas9 mRNA and sgRNA editing |
Myeloablative conditioning |
TDT |
Limited (early reports) |
< 2 years |
Increased HbF and reduced transfusion burden |
OTQ923 |
HBG1/HBG2 promoters |
Cas9 RNP, promoter disruption (HPFH-like) |
Myeloablative busulfan |
SCD |
3 (reported) |
6 - 18 months |
HbF ~ 20% - 27%; broad F-cell distribution; VOC reduction |
RM-001 |
HBG1/HBG2 promoters |
Cas9-mediated repressor site deletion |
Myeloablative busulfan |
TDT |
≥ 7 (reported) |
~ 1 - 2 years |
Rapid transfusion independence; increased total Hb |
Abbreviations: SCD, sickle cell disease; TDT, transfusion-dependent β-thalassemia; HbF, fetal hemoglobin; VOCs, vaso-occlusive crises; RNP, ribonucleoprotein; SAE, serious adverse event; HSCT, hematopoietic stem cell transplantation.
Challenges in the safety, ethical, and implementation of CRISPR/Cas9 therapy in β-hemoglobinopathies
CRISPR/Cas9-based therapies raise interconnected challenges spanning long-term genomic safety, conditioning-related toxicity, immunogenicity, and ethical and implementation considerations. However, the cellular repair outcomes are inherently heterogeneous because CRISPR/Cas9 generates DSBs at defined genomic sites. Although small insertions and deletions are often the intended consequence of NHEJ, DSBs can also produce large on-target deletions, inversions, and complex structural rearrangements at the edited locus [24]. Systematic studies in cultured and primary cells have shown that Cas9-induced DSBs may lead to deletions spanning several kilobases and chromosomal abnormalities, including chromothripsis-like events, which are not reliably detected by standard targeted amplicon sequencing approaches [24]. These structural alterations are particularly concerning in long-lived HSCs, as rare harmful events introduced at the on-target site may persist and clonally expand over time.
Off-target editing constitutes an additional and mechanistically distinct source of genomic risk beyond on-target damage. gRNAs can tolerate mismatches, allowing cleavage at partially homologous loci elsewhere in the genome [21,26]. Contemporary clinical protocols substantially mitigate off-target activity through careful gRNA design, transient delivery of Cas9 RNPs complexes, extensive preclinical screening using in silico prediction tools, in vitro assays such as GUIDE-seq or CIRCLE-seq, and targeted deep sequencing of candidate sites [10,33,42,45]. Despite these safeguards, unbiased whole-genome sequencing at sufficient depth is not routinely feasible, and low-frequency off-target or structural variants below current detection thresholds may retain long-term clinical relevance [24,25,49]. These limitations support a tiered genomic safety strategy that combines rigorous preclinical specificity assessment, focused release testing for high-risk loci, and long-term molecular surveillance after treatment [24,45,47,49].
Clonal skewing and clonal expansion constitute a third category of concern that may emerge only after extended follow-up. At the cellular level, DSBs activate DNA damage response pathways in which p53 plays a central role, triggering transient cell-cycle arrest or apoptosis in HSPCs [24,28]. Laboratory experiments have shown that Cas9 exposure can transiently favor p53-deficient or p53-dysregulated clones, raising the possibility that genome editing may amplify pre-existing premalignant populations [28]. Although no myeloid neoplasms have been conclusively attributed to CRISPR/Cas9 editing in β-hemoglobinopathy trials to date, the latency of hematologic malignancies necessitates cautious interpretation of short-term safety data. It emphasizes the need for longitudinal clonal tracking [24,45-49].
Placing these risks in historical perspective is instructive. Early γ-retroviral gene therapy trials were complicated by insertional oncogenesis, caused by enhancer-mediated activation of proto-oncogenes, leading to leukemia in several patients [6]. Lentiviral vector-based therapies, while substantially safer, have still been associated with clonal dominance and rare myeloid malignancies after long-term follow-up [6,49]. In contrast, CRISPR/Cas9-based approaches avoid random vector integration but introduce mechanistically distinct genotoxic risks related to DSB repair, including large deletions, complex rearrangements, and potential clonal selection rather than insertional mutagenesis [24,49]. From a regulatory standpoint, this distinction shifts safety monitoring toward detecting rare structural variants and long-term clonal behaviour [48,49,52].
Autologous CD34⁺ HSPCs are edited ex vivo and reinfused following myeloablative conditioning (typically busulfan) to enable engraftment [10,11,42,45,51]. Busulfan-induced myeloablation is associated with substantial acute toxicity and late complications, including infertility and secondary malignancies [5,16,48,50]. Therefore, conditioning remains a dominant contributor to overall treatment risk, particularly for young patients with SCD or β-thalassemia [3,5,48]. Consequently, alternative conditioning approaches targeting CD117 or CD45 have been investigated to deplete HSCs while sparing non-hematopoietic tissues selectively [49].
Immune responses represent an additional consideration. In healthy individuals, pre-existing antibodies and T-cell immunity against commonly used Cas9 proteins have been detected [27]. Transient Cas9 exposure during ex vivo editing and post-conditioning immunosuppression likely reduces the risk of immune-mediated rejection, and no β-hemoglobinopathy trials have reported sustained immune loss of edited cells [10,11,42,45]. Immune challenges are expected to be more prominent for future in vivo editing strategies, motivating the development of alternative nucleases and delivery systems [27,30,31].
Beyond biological safety, CRISPR/Cas9 therapies raise important ethical, economic, and implementation challenges. Genome editing for β-hemoglobinopathies is restricted to somatic cells, whereas germline editing is prohibited [13,49,52]. Key ethical issues include the need for informed consent despite long-term uncertainty, irreversible genomic modification, and justice concerns related to global access [3,11,48]. Ex vivo CRISPR/Cas9 therapies remain resource-intensive and costly, posing major barriers to their equitable deployment [16,45,48]. Therefore, long-term success depends not only on technological refinement but also on sustained regulatory oversight, ethical stewardship, and coordinated international efforts to ensure equitable access [3,47-49,52].
Critical appraisal of the current evidence and future directions
All in all, available clinical experience indicates that robust human data support short- to mid-term efficacy outcomes for ex vivo CRISPR/Cas9-based therapies, particularly for strategies targeting the erythroid-specific BCL11A enhancer or the HBG1/HBG2 promoters [10,11,42,45,50,51]. Across multiple clinical programs, these approaches have consistently achieved high rates of transfusion independence in transfusion-dependent β-thalassemia and near-complete elimination of severe vaso-occlusive crises in SCD, with follow-up extending several years in some cohorts [10,11,42,45]. Importantly, the observed safety profile in these studies is dominated by conditioning-related toxicity rather than clear adverse events attributable to genome editing itself, supporting the clinical feasibility of these platforms in highly selected patient populations [5,16,48,50].
In contrast, evidence for long-term genomic risk is derived primarily from mechanistic studies and preclinical models rather than extended clinical observation. In vitro systems, animal models, and deep sequencing analyses of edited cell products have documented on-target large deletions, chromothripsis-like rearrangements, and editing-associated clonal skewing [24,25,28]. Although these findings provide biologically plausible risk mechanisms, their true incidence and clinical relevance in humans remain uncertain, given the limited follow-up duration and the rarity of such events [24,45,49]. Conclusions regarding long-term genotoxicity should be regarded as provisional, reinforcing the need for extended post-treatment surveillance and sensitive clonal tracking in treated patients [45,48,49,52].
Similarly, concerns regarding p53-mediated selection and clonal dominance are supported by experimental data showing altered fitness landscapes in edited HSPCs [28]. However, no definitive clinical evidence has yet linked CRISPR/Cas9 editing to malignant transformation in β-hemoglobinopathy trials, and extrapolation from preclinical findings must therefore be made cautiously [10,11,42,45,49]. Historical experience with retroviral and lentiviral gene therapy underscores that such risks may only emerge after prolonged latency, highlighting the importance of long-term follow-up rather than implying established clinical harm [6,49].
Several proposed solutions discussed in this review should be interpreted in light of their evidentiary basis. Low-intensity or non-genotoxic conditioning strategies, including antibody-based approaches, are supported by encouraging preclinical and early clinical data in non-malignant settings but remain investigational in combination with genome editing [49,54]. The ability of edited HSCs to achieve durable engraftment at scale has not yet been demonstrated in large or diverse patient cohorts. Accordingly, claims regarding their impact on safety, fertility preservation, or global accessibility should be viewed as aspirational rather than established.
Likewise, in vivo genome editing represents a promising but currently speculative future direction for treating hemoglobinopathies. Although liver-directed in vivo CRISPR therapies have achieved early clinical success, efficient and selective editing of long-term repopulating HSCs in situ poses distinct biological and technical challenges [53,54]. Currently, no in vivo strategy has demonstrated durable, therapeutically sufficient modification of human HSCs, and unresolved issues related to delivery specificity, off-target effects, and immunogenicity remain substantial [27,30,31]. In this context, in vivo editing should be considered a long-term goal rather than a near-term alternative to ex vivo approaches.
Taken together, the current evidence supports a measured interpretation of progress in CRISPR/Cas9 therapy for β-hemoglobinopathies. Ex vivo genome editing strategies targeting HbF regulation are supported by compelling clinical efficacy data and manageable short-term safety profiles in specialized settings [10,11,42,45]. In contrast, long-term genomic risks, alternative conditioning strategies, and in vivo editing approaches are largely grounded in preclinical evidence and theoretical considerations [24,28,49]. Explicit differentiation between these evidence tiers is essential to avoid overstating clinical readiness and to guide responsible regulatory, ethical, and public health decision-making [48,49,52].
Access, infrastructure, and global implementation
Despite their clinical promise, current ex vivo CRISPR/Cas9 therapies for β-hemoglobinopathies are highly resource-intensive and pose substantial barriers to global implementation. These platforms require centralized cell collection and processing facilities, highly trained personnel, complex cold-chain logistics, prolonged hospitalization, and intensive supportive care surrounding myeloablative conditioning [16,45,48]. As a result, access is currently restricted to a small number of specialized centers in high-income countries, even though the greatest burden of SCD and β-thalassemia lies in low- and middle-income regions [1-4].
The development of decentralized or regional manufacturing models is one proposed strategy to improve scalability and access. Rather than relying on a limited number of centralized facilities, regional cell-processing hubs could serve multiple countries, reducing logistical complexity, transportation costs, and turnaround times. Such models require standardized manufacturing protocols, robust quality-control frameworks, and technology transfer agreements to ensure consistency and regulatory compliance across sites [45,48,49]. Early experience from HSC transplantation networks and vaccine manufacturing initiatives suggests that regionalization, coupled with workforce training and international accreditation, may offer a feasible pathway toward broader access while maintaining safety standards. Nevertheless, implementation would demand sustained investment, regulatory harmonization, and long-term partnerships between governments, academic centers, and industry.
In vivo genome editing represents a more transformative approach with the potential to alter access paradigms fundamentally. By delivering genome editors directly to HSCs within the patient, in vivo strategies could eliminate the need for ex vivo cell manipulation, specialized manufacturing facilities, and prolonged hospitalization. Advances in viral vectors, lipid nanoparticles, and antibody-targeted delivery systems have enabled in vivo CRISPR-based therapies for liver diseases in early clinical trials [53,54]. Translating this success into the hematopoietic system remains challenging, particularly in efficiently targeting long-term repopulating HSCs while avoiding off-target effects. However, in vivo editing could substantially reduce infrastructure requirements and facilitate deployment in resource-limited settings if these hurdles are overcome, thereby expanding the public health impact of genome editing for hemoglobinopathies.
Conditioning regimens represent another critical determinant of accessibility and risk. Conditioning remains a major contributor to near-term morbidity and long-term risk, and the requirement for myeloablation is a key barrier to scalability and access [5,16,48,50]. These requirements further constrain implementation in low-resource health systems. Accordingly, low-intensity or non-genotoxic conditioning strategies have emerged as a priority for improving the feasibility and acceptability of gene-editing therapies. Antibody-drug conjugates and immunotoxins targeting stem cell surface markers such as CD117 (cKit) or CD45 have shown promise in selectively depleting HSCs while sparing non-hematopoietic tissues [49,54]. If successfully combined with genome editing, such approaches could reduce hospitalisations, preserve fertility, and expand eligibility to patients currently excluded due to comorbidities or age.
These innovations underscore that the future impact of CRISPR/Cas9 therapies will depend not only on molecular efficacy but also on implementation strategies tailored to diverse health systems from a policy and public health perspective. Global initiatives for SCD and β-thalassemia emphasize that advanced therapies must be integrated alongside investments in newborn screening, infection control, hydroxyurea access, and safe transfusion practices [3,4]. To monitor long-term safety and effectiveness across populations, sustainable access will likely require a combination of tiered pricing models, outcome-based reimbursement, regional manufacturing capacity, and international registries [3,45,48,49]. Without such coordinated efforts, genome-editing therapies could widen existing health inequities rather than alleviate the global burden of β-hemoglobinopathies.
Future directions for CRISPR-based therapies in sickle cell disease and β-thalassemia
Concerns about large deletions, complex rearrangements, and p53 activation triggered by CRISPR/Cas9-mediated DSBs have spurred the development of alternative editors that can modify DNA without inducing DSBs [24,30]. Base editors pair catalytically impaired or nickase Cas9 with cytosine or adenosine deaminases to produce precise point mutations within a limited editing window [30]. In β‑hemoglobinopathies, this strategy enables correction of the sickle mutation or the addition of HPFH-like variants to HBG promoters in hemoglobinopathies [30,31,42]. Zeng et al. demonstrated that CB editors can repair β‑globin mutations in human HSPCs while preserving multilineage engraftment in xenograft models [31]. Ravi et al. generated novel HPFH-like changes in HBG promoters that increase HbF levels using base editing, laying the groundwork for future therapeutics [43]. Prime editing broadens this approach by allowing a wider array of substitutions, insertions, and deletions without the need for donor templates or DSBs [30]. Prime editing offers clear theoretical benefits for correcting the single-base HBB mutation in SCD or recreating specific HPFH alleles with minimal collateral damage, although HSPCs are still in the early preclinical stages [30,31]. The principal challenges for these next-generation editors are efficient delivery to long-term repopulating HSCs, avoidance of off-target base conversions or prime edits, and thorough analysis of bystander edits [30,31]. If these obstacles are cleared, compared with DSB-based CRISPR/Cas9, base and prime editing could lower genotoxic risk and widen the range of treatable genotypes.
Ex vivo editing requires specialized infrastructure, multiple cell manipulation steps, and myeloablation, which limit scalability and accessibility [16,43,46]. In contrast, in vivo gene editing directly delivers genome editors to HSCs in situ, potentially bypassing many of these obstacles. Recent advances in lipid nanoparticle and viral vector technologies have enabled in vivo CRISPR-based therapies for liver-targeted diseases in early-phase clinical trials (Figure 5) [51,52]. Efficient, specific delivery of HSCs to bone marrow remains challenging. Potential strategies include engineered viral vectors with HSC tropism, antibody-targeted nanoparticles that are home to stem-cell surface markers such as CD117, or transient editor expression under promoters favoring hematopoietic progenitors [30,47,53]. Early preclinical studies have demonstrated partial success in hematopoietic cell editing in vivo; however, the editing levels in bona fide long-term repopulating HSCs remain modest [51,52]. If robust in vivo HSC editing becomes feasible, it could transform the therapeutic paradigm for β‑hemoglobinopathies, allowing a finite series of infusions rather than autologous transplantation, reducing costs and enabling deployment in low-resource settings. However, in vivo approaches must address heightened concerns about systemic off-target effects, immune responses to delivery vehicles and editors, and the absence of ex vivo quality control [51,52,27].
Figure 5 Emerging strategies to improve CRISPR-based therapy delivery and conditioning. Conceptual overview of in vivo genome-editing approaches for β-hemoglobinopathies, including viral vectors, lipid nanoparticles, and targeted nanoparticles for hematopoietic stem cell delivery, together with non-genotoxic or targeted conditioning platforms that may enable reduced-intensity regimens and broaden access in future clinical applications.
The toxicity of busulfan-based myeloablation drives the overall risk of current ex vivo protocols [10,11,48,49]. New targeted conditioning regimens that selectively deplete HSCs while sparing other tissues are under development and have shown promise in human trials. Antibody-drug conjugates and immunotoxins aimed at CD117 (cKit) or CD45 can lower marrow stem cell numbers with far less systemic toxicity than conventional chemotherapy [47,54]. By combining these targeted approaches with gene editing techniques, nonmyeloablative or minimally myeloablative protocols that preserve fertility, reduce late-effect risks, and extend eligibility to older or more frail patients could be established [46,47,54]. Pairing improved conditioning with non-DSB editors could dramatically reshape the risk-benefit profile of gene editing for β‑hemoglobinopathies.
As our mechanistic understanding grows, there is increasing interest in rational combination strategies that target several facets of β‑hemoglobinopathy pathophysiology. In β‑thalassemia, this might involve pairing HbF induction with moderated α‑globin silencing to achieve an optimal globin chain balance while minimizing the risk of clinically significant α‑thalassemia [19,33,35]. Combining HbF induction with direct HBB correction or pharmacologic agents that adjust red cell hydration and adhesion could further reduce vaso-occlusion and organ injury in SCD [2,3,6,46]. Concurrently, enhanced genomic profiling of patients, including the complete spectrum of HBB mutations, co-inherited modifiers (HPFH alleles, α‑thalassemia), and background genetic risk factors, may enable a more tailored selection of editing targets and modalities [3,15–17]. Upcoming clinical trials are expected to more explicitly stratify participants by genotype and disease severity and to assess the superiority of distinct editing strategies within specific subgroups.
Future progress will not be measured solely by technological advances but also by how fully gene-editing therapies can be woven into comprehensive care models for SCD and β‑thalassemia [55]. Recent commissions have shown that parallel investment in newborn screening, infection control, hydroxyurea, safe transfusion practices, and iron chelation, together with the strategic use of advanced therapies, is essential for improving outcomes worldwide [3,4]. For gene editing, this means building scalable manufacturing platforms, including regional hubs, training multidisciplinary teams, increasing regulatory capacity in low- and middle-income countries, and developing financing mechanisms that align short-term costs with long-term health benefits [3,43,46,47]. International registries and collaborative networks are indispensable for gathering long-term safety and effectiveness data and ensuring that early adopters’ lessons guide implementation in resource-constrained settings. Ultimately, the success of CRISPR-based therapies for β‑hemoglobinopathies will be judged not only by their biological efficacy but also by their role in reducing global inequities in disease burden and access to care.
Conclusions
Since the emergence of CRISPR/Cas9, the treatment landscape for SCD and β-thalassemia has undergone rapid shifts. By focusing on the unique biology of the β‑globin locus and exploiting the critical role of HbF in dampening disease severity, gene-editing approaches have progressed from proof-of-concept studies to licensed therapies in just a decade. Ex vivo disruption of the erythroid-specific BCL11A enhancer and, more recently, editing of the HBG1/HBG2 promoters have consistently induced robust HbF expression and, in early-phase clinical trials, enabled sustained transfusion independence with total Hb levels typically in the 10 - 13 g/dL range in a majority of treated patients with transfusion-dependent β-thalassemia. At the same time, these approaches have been associated with marked reductions or absence of severe vaso-occlusive crises over follow-up periods extending several years. In SCD, these approaches have been associated with marked reductions or absence of severe vaso-occlusive crises over follow-up periods extending several years [10,11,16,33,41,43,48,49].
These clinical outcomes validate the core principles governing Hb switching and demonstrate that durable modifications to a patient’s own HSCs can provide a functional, and possibly curative, treatment for β‑hemoglobinopathies. However, they also highlight key limitations, as current protocols depend on high-dose busulfan conditioning, complex ex vivo production, and delivery at highly specialized centers whose combined constraints restrict access and increase risk, especially for children and people in low-resource settings [3,5,16,43,46]. Genomic safety, particularly the long-term effects of on- and off-target DSB repair in long-lived stem cells, remains critical and warrants rigorous investigation and continued research [24,28,43-47,50].
Future editors that do not create DSBs, along with improved conditioning methods and eventual in vivo delivery systems, offer realistic pathways toward safer, more accessible gene-editing treatments. Fulfilling this potential will require ongoing collaboration across basic researchers, clinicians, ethicists, regulators, industry, and patient groups, as well as intentional measures to ensure that advances in the field translate into fair health outcomes in regions with the greatest disease burden [3,13,46,47,50].
In summary, editing the β-globin locus with CRISPR represents a significant step forward for treating SCD and β-thalassemia. The discipline has reached a pivotal moment: Early clinical data support guarded hope, but the lasting effectiveness of these treatments will depend on meticulous risk control and a unified drive to integrate them into broad, worldwide care frameworks.
Acknowledgements
Minh Trong Quang was funded by the Master, PhD Scholarship Program of Vingroup Innovation Foundation, code VINIF.2021.ThS.69 and VINIF.2022.ThS.054.
Declaration of generative AI in scientific writing
The authors declare that no generative artificial intelligence (AI) tools were used during the preparation of this manuscript. All authors have reviewed and approved the final version and assume full responsibility for the content and conclusions of this work.
CRediT author statement
Anh Thi Ha: Conceptualization, Methodology, Literature search, Data curation, Writing - original draft, Writing - review & editing, Supervision. Nga Thi Nguyen: Conceptualization, Methodology, Validation, Writing - review & editing, Supervision. Tuan Anh Nguyen: Literature search, Data curation, Validation, Writing - review & editing. Thu-Thao Thi Huynh: Conceptualization, Methodology, Literature search, Data curation, Visualization, Writing - original draft, Writing - review & editing, Project administration, Supervision. Anh-Duy Hoang Nguyen: Literature search, Data curation, Visualization, Writing - review & editing. Minh Trong Quang: Literature search, Data curation, Visualization, Writing - review & editing.
References
[1] M Angastiniotis and S Lobitz. Thalassemias: An overview. International Journal of Neonatal Screening 2019; 5(1), 16.
[2] GJ Kato, FB Piel, CD Reid, MH Gaston, K Ohene-Frempong, L Krishnamurti, WR Smith, JA Panepinto, DJ Weatherall, FF Costa and EP Vichinsky. Sickle cell disease. Nature Reviews Disease Primers 2018; 4(1), 18010.
[3] FB Piel, DC Rees, MR DeBaun, O Nnodu, B Ranque, AA Thompson, RE Ware, MR Abboud, A Abraham, EE Ambrose, B Andemariam, R Colah, R Colombatti, N Conran, FF Costa, RM Cronin, M de Montalembert, J Elion, E Esrick, AL Greenway, IM Idris, DZ Issom, D Jain, …, K Ohene-Frempong. Defining global strategies to improve outcomes in sickle cell disease: A lancet haematology commission. The Lancet Haematology 2023; 10(8), e633-e686.
[4] A Kattamis, GL Forni, Y Aydinok and V Viprakasit. Changing patterns in the epidemiology of β-thalassemia. European Journal of Haematology 2020; 105, 692-703.
[5] MC Walters, M Patience, W Leisenring, JR Eckman, JP Scott, WC Mentzer, SC Davies, K Ohene-Frempong, F Bernaudin, DC Matthews, R Storb and KM Sullivan. Bone marrow transplantation for sickle cell disease. New England Journal of Medicine 1996; 335(6), 369-376.
[6] M Cavazzana, C Antoniani and A Miccio. Gene therapy for β-hemoglobinopathies. Molecular Therapy 2017; 25(5), 1142-1154.
[7] EB Esrick, LE Lehmann, A Biffi, M Achebe, C Brendel, MF Ciuculescu, H Daley, B MacKinnon, E Morris, A Federico, D Abriss, K Boardman, R Khelladi, K Shaw, H Negre, O Negre, S Nikiforow, J Ritz, SY Pai, …, DA Williams. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. New England Journal of Medicine 2021; 384(3), 205-215.
[8] ADH Nguyen and MT Quang. CRISPR/Cas9 genome editing in oncology: Mechanisms, therapeutic platforms and translational challenges. Molecular Biotechnology 2025. https://doi.org/10.1007/s12033-025-01533-2
[9] JB Papizan, SN Porter, A Sharma and SM Pruett-Miller. Therapeutic gene editing strategies using CRISPR-Cas9 for the β-hemoglobinopathies. Journal of Biomedical Research 2020; 35(2), 115.
[10] H Frangoul, D Altshuler, MD Cappellini, YS Chen, J Domm, BK Eustace, J Foell, J de la Fuente, S Grupp, R Handgretinger, TW Ho, A Kattamis, A Kernytsky, J Lekstrom-Himes, AM Li, F Locatelli, MY Mapara, M de Montalembert, D Rondelli, …, S Corbacioglu. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine 2021; 384(3), 252-260.
[11] B Fu, J Liao, S Chen, W Li, Q Wang, J Hu, F Yang, S Hsiao, Y Jiang, L Wang, F Chen, Y Zhang, X Wang, D Li, M Liu and Y Wu. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric β⁰/β⁰ transfusion-dependent β-thalassemia. Nature Medicine 2022; 28(8), 1573-1580.
[12] A Quagliano, D Acevedo, P Hardigan and S Prasad. Using Clustered Regularly Interspaced Short Palindromic Repeats gene editing to induce permanent expression of fetal hemoglobin in β-thalassemia and sickle cell disease: A comparative meta-analysis. Frontiers in Medicine 2022; 9, 943631.
[13] DV Parums. Editorial: First regulatory approvals for CRISPR-Cas9 therapeutic gene editing for sickle cell disease and transfusion-dependent β-thalassemia. Medical Science Monitor 2024; 30, e944204.
[14] EY Adashi, PA Gruppuso and IG Cohen. CRISPR therapy of sickle cell disease: The dawning of the gene editing era. The American Journal of Medicine 2024; 137(5), 390-392.
[15] VG Sankaran and SH Orkin. The switch from fetal to adult hemoglobin. Cold Spring Harbor Perspectives in Medicine 2013; 3(1), a011643.
[16] E Khandros and GA Blobel. Elevating fetal hemoglobin: recently discovered regulators and mechanisms. Blood 2024; 144(8), 845-852.
[17] DE Bauer and SH Orkin. Hemoglobin switching’s surprise: The versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Current Opinion in Genetics & Development 2015; 33, 62-70.
[18] N Liu, VV Hargreaves, Q Zhu, JV Kurland, J Hong, W Kim and SH Orkin. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 2018; 173(2), 430-442.
[19] S Rivella. Iron metabolism under conditions of ineffective erythropoiesis in β-thalassemia. Blood 2019; 133(1), 51-58.
[20] F Longo, A Piolatto, GB Ferrero and A Piga. Ineffective erythropoiesis in β-thalassemia: Key steps and therapeutic options. International Journal of Molecular Sciences 2021; 22(13), 7229.
[21] M Jinek, K Chylinski, I Fonfara, M Hauer, JA Doudna and E Charpentier. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337(6096), 816-821.
[22] DP Dever, RO Bak, A Reinisch, J Camarena, G Washington, CE Nicolas, M Pavel-Dinu, N Saxena, AB Wilkens, S Mantri, N Uchida, A Hendel, A Narla, R Majeti, KI Weinberg and MH Porteus. CRISPR/Cas9 β-globin gene targeting in human hematopoietic stem cells. Nature 2016; 539(7629), 384-389.
[23] MD Hoban, D Lumaquin, CY Kuo, Z Romero, J Long, M Ho, CS Young, M Mojadidi, S Fitz-Gibbon, AR Cooper, GR Lill, F Urbinati, B Campo-Fernandez, CF Bjurstrom, M Pellegrini, RP Hollis and DB Kohn. CRISPR/Cas9-mediated correction of the sickle mutation in human CD34⁺ cells. Molecular Therapy 2016; 24(9), 1561-1569.
[24] M Kosicki, K Tomberg and A Bradley. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology 2018; 36(8), 765-771.
[25] J Tycko, VE Myer and PD Hsu. Methods for optimizing CRISPR-Cas9 genome editing specificity. Molecular Cell 2016; 63(3), 355-370.
[26] RO Bak, DP Dever and MH Porteus. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nature Protocols 2018; 13(2), 358-376.
[27] CT Charlesworth, PS Deshpande, DP Dever, J Camarena, VT Lemgart, MK Cromer, CA Vakulskas, MA Collingwood, L Zhang, NM Bode, MA Behlke, B Dejene, B Cieniewicz, R Romano, BJ Lesch, N Gomez-Ospina, S Mantri, M Pavel-Dinu, KI Weinberg and MH Porteus. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nature Medicine 2019; 25(2), 249-254.
[28] OM Enache, V Rendo, M Abdusamad, D Lam, D Davison, S Pal, N Currimjee, J Hess, S Pantel, A Nag, AR Thorner, JG Doench, F Vazquez, R Beroukhim, TR Golub and U Ben-David. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nature Genetics 2020; 52(7), 662-668.
[29] MT Quang and MN Nguyen. The role and regulation of cell death in cancer. Progress in Molecular Biology and Translational Science 2025; 217, 135-161.
[30] AC Komor, YB Kim, MS Packer, JA Zuris and DR Liu. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016; 533(7603), 420-424.
[31] J Zeng, Y Wu, C Ren, J Bonanno, AH Shen, D Shea, JM Gehrke, K Clement, K Luk, Q Yao, R Kim, SA Wolfe, JP Manis, L Pinello, JK Joung and DE Bauer. Therapeutic base editing of human hematopoietic stem cells. Nature Medicine 2020; 26(4), 535-541.
[32] LC Cosenza, J Gasparello, N Romanini, M Zurlo, C Zuccato, R Gambari and A Finotti. Efficient CRISPR-Cas9-based genome editing of the β-globin gene in erythroid cells from β⁰³⁹-thalassemia patients. Molecular Therapy - Methods & Clinical Development 2021; 21, 507-523.
[33] S Mettananda, CA Fisher, D Hay, M Badat, L Quek, K Clark, P Hublitz, D Downes, J Kerry, M Gosden, J Telenius, JA Sloane-Stanley, P Faustino, A Coelho, J Doondeea, B Usukhbayar, P Sopp, JA Sharpe, JR Hughes, ..., DR Higgs. Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia. Nature Communications 2017; 8(1), 424.
[34] MC Canver, EC Smith, F Sher, L Pinello, NE Sanjana, O Shalem, DD Chen, PG Schupp, DS Vinjamur, SP Garcia, S Luc, R Kurita, Y Nakamura, Y Fujiwara, T Maeda, GC Yuan, F Zhang, SH Orkin and DE Bauer. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015; 527(7577), 192-197.
[35] G Pavani, A Fabiano, M Laurent, F Amor, E Cantelli, A Chalumeau, G Maule, A Tachtsidi, JP Concordet, A Cereseto, F Mavilio, G Ferrari, A Miccio and M Amendola. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Advances 2021; 5(5), 1137-1153.
[36] C Antoniani, V Meneghini, A Lattanzi, T Felix, O Romano, E Magrin, L Weber, G Pavani, S El Hoss, R Kurita, Y Nakamura, TJ Cradick, AS Lundberg, M Porteus, M Amendola, W El Nemer, M Cavazzana, F Mavilio and A Miccio. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood 2018; 131(17), 1960-1973
[37] N Psatha, A Reik, S Phelps, Y Zhou, D Dalas, E Yannaki, DN Levasseur, FD Urnov, MC Holmes and T Papayannopoulou. Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in β-thalassemia. Molecular Therapy - Methods & Clinical Development 2018; 10, 313-326.
[38] L Weber, G Frati, T Felix, G Hardouin, A Casini, C Wollenschlaeger, V Meneghini, C Masson, A De Cian, A Chalumeau, F Mavilio, M Amendola, I Andre-Schmutz, A Cereseto, W El Nemer, JP Concordet, C Giovannangeli, M Cavazzana and A Miccio. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Science Advances 2020; 6(7), eaay9392.
[39] EA Traxler, Y Yao, YD Wang, KJ Woodard, R Kurita, Y Nakamura, JR Hughes, RC Hardison, GA Blobel, C Li and MJ Weiss. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a benign genetic condition. Nature Medicine 2016; 22(9), 987-990.
[40] D Lu, Z Xu, Z Peng, Y Yang, B Song, Z Xiong, Z Ma, H Guan, B Chen, Y Nakamura, J Zeng, N Liu, X Sun and D Chen. Induction of fetal hemoglobin by introducing natural hereditary persistence of fetal hemoglobin mutations in the γ-globin gene promoters for genome editing therapies for β-thalassemia. Frontiers in Genetics 2022; 13, 881937.
[41] A Sharma, JJ Boelens, M Cancio, JS Hankins, P Bhad, M Azizy, A Lewandowski, X Zhao, S Chitnis, R Peddinti, Y Zheng, N Kapoor, F Ciceri, T Maclachlan, Y Yang, Y Liu, J Yuan, U Naumann, VWC Yu, …, JL LaBelle. CRISPR-Cas9 editing of the HBG1 and HBG2 promoters to treat sickle cell disease. New England Journal of Medicine 2023; 389(9), 820-832.
[42] NS Ravi, B Wienert, SK Wyman, HW Bell, A George, G Mahalingam, JT Vu, K Prasad, BP Bandlamudi and KM Mohankumar. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. eLife 2022; 11, e65421.
[43] L Wang, H Xu, J Liang, Y Li, L Shi, B Xiao, J Huang, X Xu and X Zhang. Initial safety and efficacy study of RM-001 autologous HBG1/2 promoter-modified CD34⁺ cells in transfusion-dependent β-thalassemia. HemaSphere 2022; 6, 1347-1348.
[44] R Liu, L Wang, H Xu, X Yin, J Liang, W Xie, G Yang, Y Li, Y Zhou, L Shi, B Xiao, L Shi, Z Shi, X Zhou, J Fang, X Xu, Y Lai, J Huang and X Zhang. Safety and efficacy of RM-001 in transfusion-dependent β-thalassemia: Early results. HemaSphere 2023; 7(S3), e613965e.
[45] M Laurent, M Geoffroy, G Pavani and S Guiraud. CRISPR-based gene therapies: From preclinical to clinical treatments. Cells 2024; 13(10), 800.
[46] L Li and PK Mandal. Recent. Recent advancements in gene therapy for sickle cell disease and β-thalassemia. Frontiers in Hematology 2024; 3, 1468952.
[47] A Aprile, MR Lidonnici and G Ferrari. Gene therapy for hemoglobinopathies: Clinical trial results and biology of hematopoietic stem cell and the bone marrow niche. Cell Reports Medicine 2025; 6(12), 102419.
[48] H Frangoul, F Locatelli, A Sharma, M Bhatia, M Mapara, L Molinari, D Wall, RI Liem, P Telfer, AJ Shah, M Cavazzana, S Corbacioglu, D Rondelli, R Meisel, L Dedeken, S Lobitz, M de Montalembert, MH Steinberg, MC Walters, …, SA Grupp. Exagamglogene autotemcel for severe sickle cell disease. New England Journal of Medicine 2024; 390(18), 1649-1662.
[49] F Locatelli, P Lang, D Wall, R Meisel, S Corbacioglu, AM Li, J de la Fuente, AJ Shah, B Carpenter, JL Kwiatkowski, M Mapara, RI Liem, MD Cappellini, M Algeri, A Kattamis, S Sheth, S Grupp, R Handgretinger, P Kohli, ..., H Frangoul. Exagamglogene autotemcel for transfusion-dependent β -thalassemia. New England Journal of Medicine 2024; 390(18), 1663-1676.
[50] R Handgretinger and M Mezger. An evaluation of exagamglogene autotemcel for the treatment of sickle cell disease and transfusion-dependent beta-thalassaemia. Expert Opinion on Biological Therapy 2024; 24(9), 883-888.
[51] JD Finn, AR Smith, MC Patel, L Shaw, MR Youniss, J van Heteren, T Dirstine, C Ciullo, R Lescarbeau, J Seitzer, RR Shah, A Shah, D Ling, J Growe, M Pink, E Rohde, KM Wood, WE Salomon, WF Harrington, ..., DV Morrissey. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Reports 2018; 22(9), 2227-2235.
[52] JD Gillmore, E Gane, J Taubel, J Kao, M Fontana, ML Maitland, J Seitzer, D O’Connell, KR Walsh, K Wood, J Phillips, Y Xu, A Amaral, AP Boyd, JE Cehelsky, MD McKee, A Schiermeier, O Harari, A Murphy, ..., D Lebwohl. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. New England Journal of Medicine 2021; 385(6), 493-502.
[53] B Fu, X Zhang, L Wang, J Liao, S Chen, B Zheng, W Li, F Wang, D Li, M Liu and Y Wu. Updated follow-up of BRL-101 CRISPR-Cas9-mediated BCL11A enhancer editing for transfusion-dependent β-thalassemia. HemaSphere 2023; 7(S3), e406095b.
[54] A Czechowicz, R Palchaudhuri, A Scheck, Y Hu, J Hoggatt, B Saez, WW Pang, MK Mansour, TA Tate, YY Chan, E Walck, G Wernig, JA Shizuru, F Winau, DT Scadden and DJ Rossi. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nature Communications 2019; 10(1), 617.
[55] TTT Huynh, TN Nguyen, TA Nguyen, ADH Nguyen and MT Quang. Convergence of gene therapy and vaccine platforms in the post-pandemic era: A mini review. Trends in Sciences 2026; 23(3), 12197.