Trends
Sci.
2026;
23(8):
12603
The Influence of Para-Substituent on the Molecular Properties and Biological Activities of Aroylhydrazone Derivatives: DFT Analysis and Anti-Cervical Cancer Evaluation Through In Silico and In Vitro Studies
Imanuel
Gauru1,2,
![]() ,
Yusuf Syahril Alam1,
,
Yusuf Syahril Alam1,
![]() ,
Syafri Izzat Abidiy1,
,
Syafri Izzat Abidiy1,
![]() ,
,
Fidelis
Nitti2,
![]() ,
Holilah Holilah3,
,
Holilah Holilah3,
![]() ,
Yuly Kusumawati1,
,
Yuly Kusumawati1,
![]() ,
Mardi Santoso1,
,
Mardi Santoso1,
![]() ,
,
Arif
Fadlan1,
![]() and Fahimah Martak1,*,
and Fahimah Martak1,*,
![]()
1Department of Chemistry, Faculty of Science and Data Analytics, Sepuluh Nopember Institute of Technology,
Surabaya 60111, Indonesia
2Department of Chemistry, Faculty of Science and Engineering, University of Nusa Cendana, Kupang 85001, Indonesia
3Research Center for Biomass and Bioproducts, National Research and Innovation Agency of Indonesia (BRIN),
Cibinong 16911, Indonesia
(*Corresponding author’s e-mail: [email protected])
Received: 9 November 2025, Revised: 29 December 2025, Accepted: 5 January 2026, Published: 15 March 2026
Abstract
The synthesis of four aroylhydrazone-derived compounds was successfully conducted via the condensation reaction between benzoyl hydrazine and four different aromatic aldehydes. The resulting comppunds were identified as 4-Methoxybenzylidene ben-zo hydrazide (L1), 4-hidroksi-benzaldehid benzoyl hydrazone (L2), 4-methylbenzylidene benzohydrazide (L3) and 4-nitrobenziliden - 2 - benzoyl hydrazine (L4). Their spectroscopi cal and physicochemical characteristics were analyzed through UV-Vis, IR, 1H NMR, and 13C NMR instruments. Structural optimization was subsequently performed using density functional theory (DFT), followed by theoretical evaluations of UV-Vis and IR spectral modeling, reactivity and stability, and several quantum chemical descriptors. It was revealed that the theoretical results agreed with the experimental yields. The results of DFT analysis showed that L1 (ΔEGAP = 4.58 eV) and L3 (ΔEGAP = 4.42 eV) have high molecular stability and reactivity. These parameters indicate L1 and L3 as candidates for anticancer compounds. Besides, the antioxidant and cytotoxicity of the prepared compounds were observed to elucidate their specific mechanisms. L1 and L3 showed moderate anticancer activity with IC50 values of 82.12 and 42.64 µg/mL, respectively. In addition, insilico analysis, indicated strong binding affinities for L1 and L4, with docking binding energies of −9.37 and −10.57 kcal/mol. The Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) predictions and pharmacokinetic parameters of the samples further supported their favorable oral bioavailability.
Keywords: Aroylhydrazone, Characterization, Density functional theory, ADMET, Molecular docking
Introduction
Aroylhydrazones, a subclass of azomethine, are distinguished by the existence of the –C=N–N– group and are typically synthesized through condensation reactions between hydrazide and aldehyde or ketone [1]. These compounds possess key donor sites, notably carbonyl (–C=O) and azomethine (–C=N–), which facilitate interaction with various biological targets. In addition, structural modification through the addition of
various substituents on the benzene ring has been shown to enhance their biological activity profiles significantly. Currently, there has been a rising emphasis on advancing aroylhydrazone-derived compounds. Notably, those based on ((E) - N’- benzylidenebenzohydrazide) Ar-CH=N–NH–C (=O) –Ar′ (where Ar′ represents an aromatic ring) have garnered particular interest due to their superior chemical stability and broader synthetic versatility compared to their alkyl-substituted hydrazone counterparts [2]. Consequently, structural features position them as promising scaffolds for further design and development of bioactive and pharmaceutical agents. Numerous studies have exhibited that aroylhydrazones possess the ability to inhibit metalloproteins and disrupt intracellular homeostasis, primarily by interacting with DNA [3]. Moreover, aroyl hydrazone-based compounds have been reported to exhibit extensive pharmacological activities, such as anti-inflammatory [4], anti-microbial [5], anticonvulsant [6], anti-tubercular [7], anti-tumor [8], anti-fungal, and anti-viral effects [9]. Notably, structural modification at the para position of the aromatic ring has been shown to significantly impact the compound’s lipophilicity, electronic distribution, and steric profile, as shown in Figure 1 [10]. Despite significant interest in aroylhydrazone recently, relatively limited studies have examined the structure-activity relationships of para-substituted benzaldehyde-based aroylhydrazones, particularly with respect to their antioxidant and anticancer activities, employing a combination of molecular docking and computational chemistry approaches. Therefore, the current investigation aims at designing and synthesizing four aroylhydrazone derivatives, employing electron-donating and electron-withdrawing substituents, namely methoxy, hydroxy, methyl, and nitro groups at the para-position of the benzaldehyde moiety. These substituents are anticipated to significantly influence several physicochemical properties of the compounds, such as chemical reactivity, lipophilicity, molecular polarity, hydrophobic interactions, and, ultimately, biological efficacy. To assess the bioactivity of the synthesized compounds initially, both in vitro and in silico observations were employed. In this regard, computational chemistry serves as a crucial approach by facilitating the understanding of chemical reactivity and the establishment of structure-reactivity correlations. It also allows for the estimation of vibrational, electronic, and thermodynamic characteristics of the compounds examined in environments of biological and chemical significance [11]. The results obtained from this approach may be applied as additional support for the analytical and experimental findings [12]. Among the computational approaches, DFT is mostly employed due to its widespread application and reliability in quantum chemical modeling, particularly for calculating various molecular properties such as molecular structure and biological activity. This method enables the determination of molecular structures and relative energies with enhanced accuracy by using an appropriate basis set, surpassing the limitations of conventional ab initio methods such as Restricted Hartree-Fock (RHF) and second-order Moller-Plesset Perturbation Theory (MP2) [13]. In this study, four aroylhydrazone derivatives (L1 - L4) were rationally designed and synthesized from para-substituted benzaldehyde and benzohydrazide. Their structures were elucidated through spectroscopic techniques, DFT, and molecular docking analyses. They were also systematically examined with diverse spectroscopic techniques, including Fourier-transform infrared spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance (1H and 13C NMR), and high-resolution mass spectrometry (HRMS). Quantum mechanical investigations were performed using DFT calculations, including molecular geometry optimizations, electrostatic potential mapping with atomic charge distributions, frontier molecular orbital (HOMO-LUMO) energy calculations, and evaluation of thermodynamic parameters. Dipole moment values for the prepared compounds were also computed employing the B3LYP method with the 6-31G* basis set. Additionally, the in vitro biological activities of the compounds were assessed through antioxidant assays and cytotoxicity testing against HeLa cervical cancer cells. The interaction mechanisms between the synthesized compounds (L1 - L4) and target biomolecules in HeLa cells were studied through molecular docking analysis.
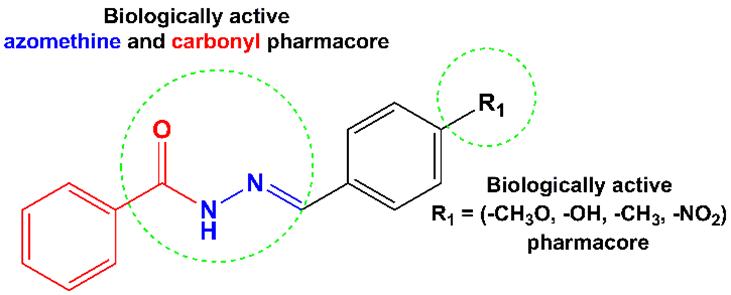
Figure 1 Designed (E)-N’-benzylidenebenzohydrazide.
Materials and methods
Materials
All starting materials and solvents used in this work, including benzoyl hydrazine, various aldehydes, and PrestoBlue reagent, were of analytical grade and purchased from Sigma-Aldrich, and utilized directly in reactions without purification.
General procedure for the synthesis of aroylhydrazone derivatives
Synthesis of 4-Methoxybenzylidene benzo hydrazide (L1)
A mixture of 6 mmol of benzoyl hydrazine (0.82 g) and 6 mmol of 4-methoxybenzaldehyde (0.73 mL) was refluxed in 40 mL of ethanol at 95 °C for 3 h. Afterward, glacial acetic acid (6 drops) was added to the mixture to catalyze the reaction. The reaction was supervised with thin-layer chromatography (TLC) employing the solution of n-hexane and ethyl acetate (1:1 volume ratio). After refluxing, it was left to reach room temperature so that the compound could undergo crystallization. Further, it was cleaned thoroughly with ethanol, producing the final product: 85.45%, white purified aroylhydrazone solid. Molecular Formula: C15H14N2O2, M.p. 179 ⁰C, UV-Vis (DMSO) λmax (nm): 320, IR (KBr, ʋ (cm-1)): 1170 (CO), 1307 (C–N), 1606 (C=N), 1649 (C=O), 2841 (Csp3–H), 3255 (Csp2–H), 3421 (N–H). 1H NMR (500 MHz, DMSO) δ(ppm) 3.83 (s, 3H, -CH3), 6.97 - 6.99 (dd, 2H, Ar H), 7.52 (t, 2H, Ar H), 7.59 - 7.62 (m, 1H, Ar H), 7.77 - 7.80 (dd, 2H, Ar H), 7.93 -7.95 (dd, 2H, Ar H), 8.29 (s, 1H, -CH); 13C NMR (126 MHz, DMSO) δ(ppm) 54.46 (-CH3), 113.85 (Ar C), 126.62 (Ar C), 127.33 (Ar C), 128.35 (Ar C), 129.15 (Ar C), 131.85 (Ar C), 132.82 (Ar C), 149.23 (–CH), 161.86 (Ar C), 165.54 (–C=O). HRMS (ESI-TOF) calculated for C15H14N2O2 [M+Na]+ 277.095, found 277.094.
Synthesis of 4-Hydroxy-benzaldehyde benzoyl hydrazone (L2)
Benzoyl hydrazine (6 mmol, 0.82 g) and 4-hydroxybenzaldehyde (6 mmol, 0.73 g) were refluxed in 40 mL of ethanol at 95⁰ °C for 3 h. Similarly, the reaction was catalyzed with glacial acetic acid and tracked using TLC. After refluxing, it was left to reach room temperature so that the compound could undergo crystallization. Further, it was cleaned thoroughly with ethanol, resulting in the final product: 36.64%, white purified aroyl hydrazone solid. Molecular Formula: C14H12N2O2, M.p. 279 °C, UV-Vis (DMSO) λmax (nm): 291, IR (KBr, ʋ (cm−1)): 1305 (C–O), 1342 (C–N), 1784 (C=N), 1647 (C=O), 3016 (Csp2–H), 3462 (N–H), 3180 (–OH). 1H NMR (500 MHz, DMSO) δ(ppm) 6.84 - 6.87 (dd, 2H, Ar H), 7.53 (t, 2H, Ar H), 7.59 - 7.63 (m, 1H, Ar H), 7.69 - 7.72 (dd, 2H, Ar H), 7.93 - 7.95 (dd, 2H, Ar H), 8.26 (s, 1H, –CH); 13C NMR (126 MHz, DMSO) δ(ppm) 115.25 (Ar C), 125.39 (Ar C), 127.29 (Ar C), 128.34 (Ar C), 129.33 (Ar C), 131.80 (Ar C), 132.88 (Ar C), 149.65 (–CH), 159.99 (Ar C), 165.54 (–C=O). HRMS (ESI-TOF) calculated for C14H12N2O2 [M+Na]+ 263.08, found 263.07.
Synthesis of 4-methylbenzylidene benzohydrazide (L3)
Benzoyl hydrazine (6 mmol, 0.82 g) and 4-methylbenzaldehyde (6 mmol, 0.70 mL) were refluxed in 40 mL of ethanol at 95 °C for 3 h. Similarly, the reaction was catalyzed with glacial acetic acid and tracked using TLC. After refluxing, it was left to reach room temperature so that the crystalline compound could be obtained. Further, it was rinsed thoroughly with ethanol, resulting in the final yield: 44.79%, white purified aroyl hydrazone solid. Molecular Formula: C15H14N2O, M.p. 193 °C, UV-Vis (DMSO) λmax (nm): 307, IR (KBr, ʋ (cm−1)): 1062 (C–O), 1776 (C=N), 1641 (C=O), 3051 (Csp2–H), 3028 (Csp3–H), 3394 (N–H). 1H NMR (500 MHz, DMSO) δ(ppm) 2.38 (s, 3H, –CH3), 7.25 (d, 2H, Ar H), 7.51 (t, 2H, Ar H), 7.5 9 - 7.72 (dd, 2H, Ar H), 7.93 - 7.96 (dd, 2H, Ar H), 8.32 (s, 1H, –CH); 13C NMR (126 MHz, DMSO) δ(ppm) 20.17 (–CH3), 127.37 (Ar C), 127.49 (Ar C), 128.36 (Ar C), 129.10 (Ar C), 131.38 (Ar C), 131.91 (Ar C), 132.76 (–CH), 140.84 (Ar C), 149.42 (–CH), 165.62 (–C=O). HRMS (ESI - TOF) calculated for C15H14N2O [L+Na]+ 261.27, found 261.10.
Synthesis of 4-Nitrobenzylidene-benzoyl hydrazine (L4)
Benzoyl hydrazine (6 mmol, 0.82 g) and 4-nitrobenzaldehyde (6 mmol, 0.82 g) were refluxed in 40 mL of ethanol at 95⁰ °C for 3 h. Similarly, the reaction was catalyzed with glacial acetic acid, and the progress was tracked using TLC. After refluxing, it was left to reach room temperature so that the crystalline compound could be obtained. Further, it was rinsed thoroughly with ethanol, resulting in the final yield: 94.44%, yellow purified aroyl hydrazone solid. Molecular formula: C14H11N3O3, M.p. 265 °C, UV-Vis (DMSO) λmax (nm): 342, IR (KBr, ʋ (cm−1)): 1751 (C=N), 1604 (C=O), 3059 (Csp2-H), 3269 (N–H), 1550 (N=O). 1H NMR (500 MHz, DMSO) δ(ppm) 7.56 (t, 2H, Ar H), 7.63 (t, 1H, Ar H), 7.94 (d, 2H, Ar H), 8.013 (d, 2H, Ar H), 8.32 (d, 2H, Ar H), 8.56 (s, 1H, –CH), 12.20 (s, 1H, –NH); 13C NMR (126 MHz, DMSO) δ(ppm) 123.52 (Ar C), 127.14 (Ar C), 127.42 (Ar C), 127.97 (Ar C), 131.49 (Ar C), 132.41 (Ar C), 140.03 (Ar C), 144.62 (–CH), 147.23 (Ar C), 162.80(–C=O). HRMS (ESI-TOF) calculated for C14H11N3O3 [L+Na]+ 292.07, found 292.07.
Characterization
The synthesized molecules were analyzed using various instruments, including UV-Vis spectrophotometer Genesys 10S (Thermo Fisher Scientific, Madison, WI, USA), FTIR Shimadzu 8400S (Tokyo, Japan), and NMR Agilent 500 MHz fitted with a DD2 console, functioning at 500 MHz for proton (¹H) and 125 MHz for carbon (¹³C) analysis. High-resolution mass spectra were obtained using a Waters Q-TOF MS Xevo mass spectrometer (USA).
Cytotoxicity test
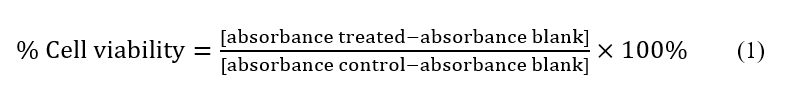
The cytotoxic activity toward HeLa cancer cells (human cervical adenocarcinoma) was evaluated in vitro employing the PrestoBlue (PB) assay method [14]. For this procedure, RPMI medium consisting of fetal bovine serum (FBS) (10 %(v/v)) and antibiotics (50 µL/50 mL) were used to culture the cells. The cultured cells (170,000 cells/mL) were then plated into 96-well microplates and kept for 24 h at 37 °C with 5% CO₂ in a humidified chamber. Following incubation, the existing culture medium was substituted with new medium consisting of diverse concentrations of the test compounds (7.81, 15.63, 31.25, 62.50, 125, 250, 500, and 1000 µg/mL). The treated cells were subsequently incubated for 48 h. Afterward, PrestoBlue reagent (Thermo Fisher Scientific, Uppsala, Sweden) was introduced into each well, and the absorbance was investigated at 570 nm via a multimode plate reader. Cisplatin was utilized as the positive control to compute the half-maximal inhibitory concentrations (IC50) using linear regression. To calculate the percentage of cell viability using the following Eq. (1) [15]:
Antioxidant activity assay
The antioxidant activity of the prepared aroyl hydrazones was examined with the 2,2-diphenyl-1-picrylhydrazyl (DPPH) free-radical-scavenging assay [16]. Gallic acid and quercetin served as reference antioxidants, while dimethyl sulfoxide (DMSO) acted as the solvent control. To perform the test, 1 mL of DPPH (6×10−5 M) was combined with 33 µL of the sample solution dissolved in DMSO at diverse concentrations of 319.46, 159.73, 79.87, 39.93, 19.97, 9.98, 4.99, 2.50, and 1.25 g/mL. Each mixture was preserved in a 37 °C water bath for 30 min, and the absorbance (S) at 517 nm was measured using a UV-Vis spectrophotometer. The blank solution (B) consisted of 33 µL of DMSO plus DPPH solution. The radical-scavenging activity of DPPH was determined using the formula of [(B-S)/B]×100% [17].
ADMET
The drug-likeness and ADMET of all samples were assessed with the ADMET Lab 2.0 software. The assessment of the drug-likeness followed Lipinski’s Rule of 5, considering main physicochemical aspects of molecular weight, partition coefficient (log D), and the number of hydrogen bond donors and acceptors. Meanwhile, the ADMET prediction offered comprehensive data regarding the pharmacokinetic aspects of each compound, including intestinal absorption, distribution characteristics, metabolic stability, excretion potential, and toxicity risks [18].
DFT calculations
The DFT calculations were done using the Gaussian 16 software to obtain molecular descriptors for the 4 hydrazone compounds. Geometry optimizations were evaluated via the B3LYP method with the 6-31G* basis set to ensure that each structure reached its minimum energy conformation. The frequency calculation was carried out to predict their IR vibrational modes, while electronic spectra were computed to estimate the maximum UV-Vis absorption wavelength [19]. Frontiers molecular orbital energies (HOMO and LUMO) were employed to estimate several important quantum chemical descriptors, such as energy band gap (ΔEGAP), global hardness (η), global softness (σ), absolute electronegativity (χ), electrophilicity index (ω), and dipole moment (μ). These parameters were calculated using Eqs. (2) - (7) [20].

Molecular docking analysis
The Cyclin Dependent Kinase 2 (CDK2) with PDB ID: 5IEV was selected as the target receptor for molecular docking studies involving the aroyl hydrazone-derived ligands (L1 - L4). The protein preparation involved the elimination of the crystallographic water portion and unnecessary co-crystallized ligands. All missing atoms were repaired, and polar hydrogens and Kollman charges were added to activate the protein structure docking. The ligand structures (L1 - L4) were prepared and incorporated into the molecular docking system. The docking protocol was confirmed using a self-docking (re-docking) procedure, where the native co-crystallized ligand (R0N) was re-docked into the active site of CDK2. The resulting root-mean-square-deviation (RMSD) obtained was 1.20 Å, confirming the reliability of the docking setup [21]. Grid box dimensions were set to 40×40×40 with a spacing of 0.5, and the grid center was arranged at the Cartesian coordinates of ‒11.057, ‒9.3, and ‒10.687, respectively. Docking parameters were configured using the Lamarckian Genetic Algorithm with a population size of 300 and 100 genetic algorithm runs. All docking procedures were performed with AutoDock Tools version 1.5.7. The resulting ligand-protein complexes were subsequently analyzed and visualized in 2 dimensions using Biovia Discovery Studio to evaluate ligand-protein interactions in 2D [22].
Results and discussion
Chemical structures
Aroylhydrazone derivatives derived from para-benzaldehyde and borohydride were successfully synthesized via the modified reflux method, conducted for 3 h [23]. The synthesis involved an equimolar (1:1) ratio of the starting materials and the catalyst (glacial acetic acid). The general reaction mechanism to form aroyl hydrazone derivatives is visualized in Figure 2, where water molecules are released as by-products. The reaction begins with a nucleophilic attack by the nitrogen atom of benzoyl hydrazine, containing a lone electron pair, on the electrophilic carbon of the aldehyde’s carbonyl group. This stage generates an intermediate facilitated by a proton transfer process [24]. Subsequent proton transfer from nitrogen to oxygen, mediated by glacial acetic acid, enables the formation of an azomethine linkage. The acetate ion then promotes an elimination reaction, forming an azomethine group and releasing water molecules [25]. TLC tests were performed to validate the formation of the resulting desired compounds and assess their purity [26].
UV-Vis spectra analysis
The UV-Vis absorption spectra of aroyl hydrazone compounds (L1 - L4), obtained from the experiment and TD DFT calculations in DMSO solvent, indicate that the absorption bands are predominantly located in the ultraviolet region (Figure 3). Experimentally, the maximum absorption wavelengths were recorded at 315 nm for L1, 325 nm for L2, 305 nm for L3, and 342 nm for L4. In comparison, the calculated spectra showed absorption maxima at 311 nm (L1), 324 nm (L2), 313 nm (L3), and 323 nm (L4). The absorption band within the wavelength range of 300 - 373 nm is primarily ascribed to the n → π∗ electronic transitions involving the imine (C=N) and (N‒NH) groups, where electrons are excited from non-bonding orbitals (n) to anti-bonding orbitals (π*) [27,28]. Meanwhile, the bands at shorter wavelengths, specifically at 240 and 267 nm for L1; 237, 240, 271, 273 nm for L2; 234, 244, 265 nm for L3, and 278 nm for L4, are primarily associated with the π→ π∗ transitions of the aromatic C=C group of the benzene rings.
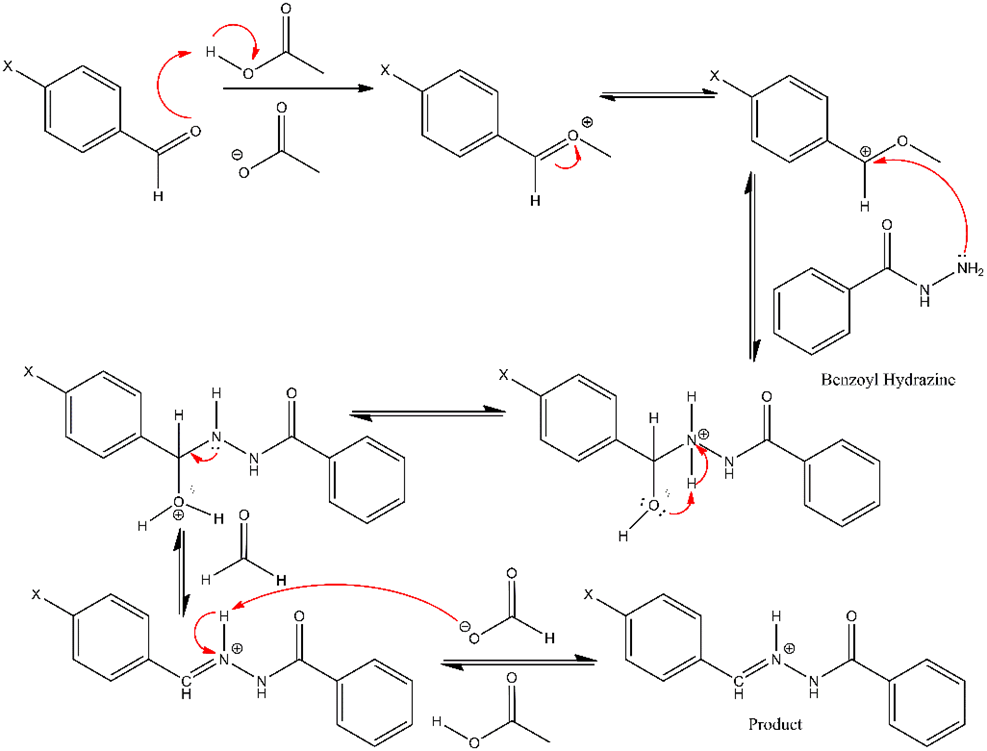
Figure 2 The reaction mechanism for the formation of aroylhydrazone derivatives from para- benzaldehyde and benzohydrazide.
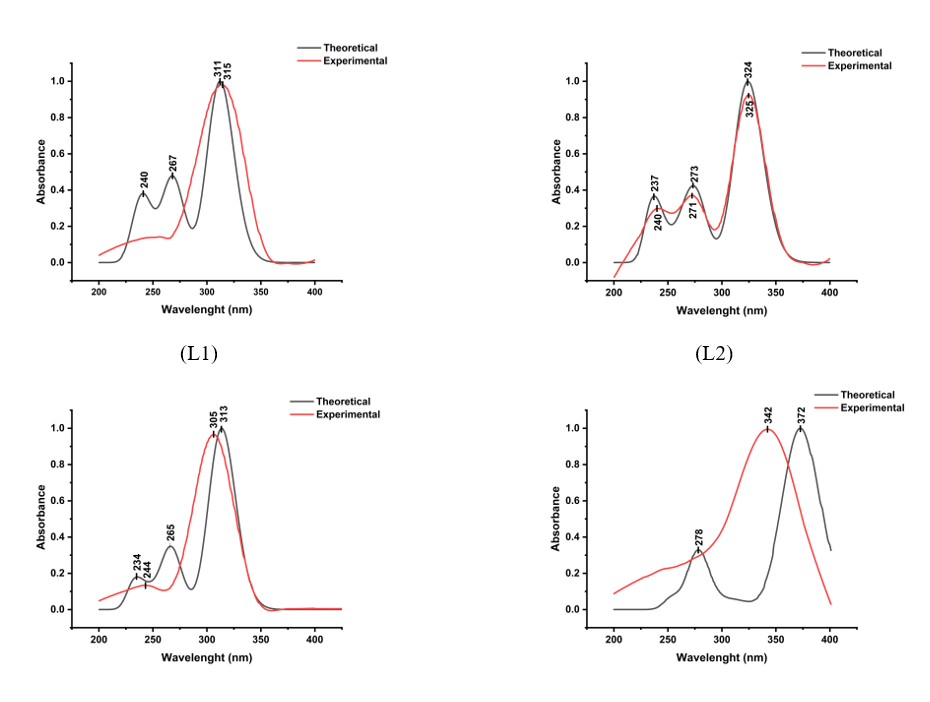
Figure 3 The theoretical and experimental UV-Vis spectra of all samples.
FTIR spectra analysis
The FTIR spectra of all samples obtained experimentally and theoretically are presented in Table 1 summarizes the vibrational frequencies and denotes the vibration modes of the identified functional groups. The data exhibited that the theoretical frequencies agreed well with the experimentally observed frequencies. The variation in vibrational modes observed in the experimental spectra of the aroyl hydrazone derivatives of para-benzaldehyde and benzo hydrazide can be explained as follows:
4-Methoxybenzylidene benzohydrazide (L1)
Based on Table 1, the absorption peaks at 3,421 and 3,255 cm−¹ denote the N–H and Csp²–H groups, respectively. The low peak observed at 2,841 cm−¹ can be assigned to the aromatic group of Csp³–H. The peak at 1,606 cm−¹ represents the C=O vibration. The prominent peak at 1,514 cm⁻¹ was related to the imine group (C=N), which is characteristic of compound L1 [29,30]. Additionally, the peak in the 1,050 - 1,320 cm⁻¹ range could be associated with the existence of the C–O group. The peaks detected in the 1,020 - 1,250 cm−¹ range denoted the C–N functional group. Finally, the significant and sharp peak at 713 cm−¹ denoted the existence of the C–H group.
4-Hydroxy-benzaldehyde benzoyl hydrazine (L2)
The reasonable intensity peaks observed at 3,269 and 3,198 cm−1 were related to the N–H and O–H vibrations, respectively. The weak absorption peak at 3,030 cm−1 matched the aromatic group Csp2–H. Meanwhile, the peak at 2,833 cm−1 could be linked to the Csp3–H aliphatic group. The peak at 1,604 cm−1 emerged primarily due to the presence of the C=O functional group. The distinct and highly intense peak observed at 1,514 cm−1, characteristic of compound L2, was attributed to the hydrazone group (C=N–N) [31]. Besides, the peaks observed in the regions of 1,250 - 1,310 cm−1 and 1,020 - 1,250 cm−1 indicated the C–O and C–N functional groups, respectively. Finally, the sharp and intense peak at 837 cm−1 was attributed to the C–H group.
4 - Metilbenzelidene – 2 – benzoyl hydrazine (L3)
The weak absorption at 3,200 and 3,051 cm−1 denoted the N–H and Csp–H vibrations, respectively. Besides, the peaks at 3,028 and 1,641 cm−1 represented the Csp3-H aromatic and C=O, respectively. Generally, the C = N vibrations emerge in the range of 1,600 - 2,000 cm−1. The strong and typical peak located at 1,602 cm−1 corresponded to the C=N imine group, which is characteristic of compound L3. In addition, the band observed in the 1,020 - 1,250 cm−1 range was associated with the C–N vibration. Finally, the peak at 773 cm−1 was linked to the C–H group.
4-Nitrobenzylidene – benzoyl hydrazine (L4)
The absorption peaks observed at 3,180 and 3,016 cm−1 are ascribed to the vibrations of N – H and Csp2-H groups, respectively. A low intensity band at 2,839 cm−1 was associated with the vibration mode of aromatic Csp3-H groups. The peak at 1,647 cm−1 denoted the vibration mode of the C = O group. A strong peak at 1,566 cm−1 was linked to imine C=N groups, representing the characteristic peak of compound L4. Besides, the peak at 1,521 cm−1 was related to the N–O groups. Finally, the peak at 698 cm−1 matches the out-of-plane bending vibration of the C–H group. The linear correlation between the theoretical and experimental FTIR spectra is visualized in Figure 4. In general, a linear trend was observed, with correlation coefficients (R2) of 0.994 for L1, 0.985 for L2, 0.995 for L3, and 0.994 for L4. The R2 values close to 1.00 confirm the strong alignment between experimental yields and theoretical calculations, indicating the computational scaling strategy is highly reliable [32].
Table 1 FTIR data of all samples (experimental and theoretical).
Compounds |
Experimental (cm−1) |
Theoretical (cm−1) |
Assignment |
L1
|
713 |
727 |
ΡOUT(C–H) |
1,170 |
1,211 |
ʋ(C – N) |
|
1,261 |
1,307 |
ʋ(C – O) |
|
1,514 |
1,664 |
ʋ(C = N) |
|
1,606 |
1,757 |
ʋ(C = O) |
|
2,841 |
3,031 |
ʋ(Csp3 – H) |
|
3,255 |
3,206 |
ʋ(Csp2 – H) |
|
3,421 |
3,484 |
ʋ(N – H) |
|
L2
|
837 |
719 |
ΡOUT(C–H) |
1,166 |
1,211 |
ʋ(C – N) |
|
1,282 |
1,314 |
ʋ(C – O) |
|
1,512 |
1,668 |
ʋ(C = N) |
|
1,604 |
1,757 |
ʋ(C = O) |
|
2,833 |
3,043 |
ʋ(Csp3 – H) |
|
3,030 |
3,194 |
ʋ(Csp2 – H) |
|
3,198 |
3,206 |
ʋ(-OH) |
|
3,269 |
3,747 |
ʋ(N – H) |
|
L3
|
773 |
720 |
ΡOUT(C–H) |
1,138 |
1,189 |
ʋ(C – N) |
|
1,284 |
1,335 |
ʋ(C – O) |
|
1,602 |
1,518 |
ʋ(C = N) |
|
1,641 |
1,759 |
ʋ(C = O) |
|
3,028 |
3,042 |
ʋ(Csp3 – H) |
|
3,051 |
3,045 |
ʋ(Csp2 – H) |
|
3,200 |
3,206 |
ʋ(N – H) |
|
L4
|
698 |
722 |
ΡOUT(C–H) |
1,151 |
1,130 |
ʋ(C – N) |
|
1,342 |
1,324 |
ʋ(C – O) |
|
1,566 |
1,648 |
ʋ(C = N) |
|
1,647 |
1,662 |
ʋ(C = O) |
|
2,839 |
1,768 |
ʋ(Csp3 – H) |
|
3,016 |
3,062 |
ʋ(Csp2 – H) |
|
3,180 |
3,209 |
ʋ(N – H) |
|
1,521 |
1,375 |
ʋ(N – O) |
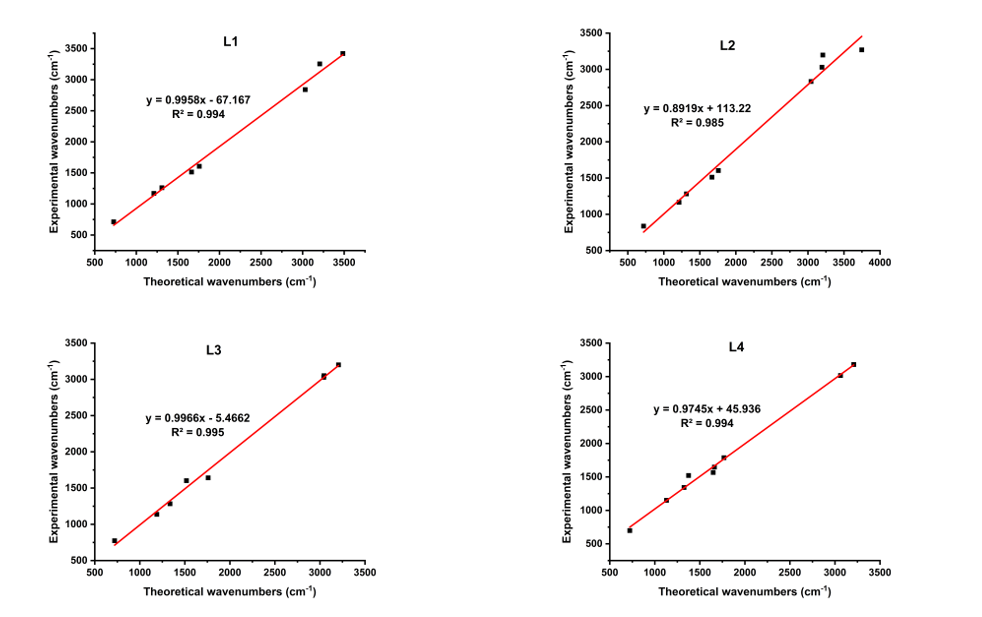
Figure 4 Correlation curves between the theoretical and experimental wavenumbers of all samples.
NMR analysis
The NMR spectral analysis of compounds L1 - L4, conducted at room temperature in DMSO-d6, provided valuable insights into their structural features, as summarized in Table 2. The 1H NMR spectra for all four compounds revealed characteristic aromatic proton signals, appearing at chemical shifts (δ) range of 6.85 - 8.32 ppm, indicative of both benzene and heterocyclic ring environments (CHAr - benzene, and CHAr - heterocyclic). The hydrazone group (CH=N–N) was identified by distinct singlet signals at δ 8.45 (L1), 8.36 (L2), 8.43 (L3), and 8.56 ppm (L4), aligning with literature values. Additionally, signals at δ 11.77 (L1), 11.65 (L2), 11.79 (L3), and 12.20 ppm (L4) correspond to the protons of the (NH–N–) group [33]. Methyl (–CH₃) group protons in L3 and L1 showed chemical shifts at 2.40 ppm and 3.85 ppm, respectively, while L2 presented a broadened signal at 9.95 ppm for the hydroxyl (–OH) proton. The ¹³C NMR spectra of L1, L2, L3, and L4 exhibited signals at δ 149.23, 149.45, 149.42, and 144.62 ppm, attributed to the imine group (–N=CH–) [34]. Each compound displayed a peak at 165.54 ppm for L1 and L2, 165.62 ppm for L3, and 162.80 ppm for L4, corresponding to the carboxylic acid group (COOH). Aromatic ring carbon signals were observed between δ 113.85 and 140.84 ppm. Chemical shifts at δ 54.6 ppm for L1 and 20.17 ppm for L3 indicated carbon atoms in the methyl group (-CH₃).
Table 2 1H NMR and 13C NMR chemical shifts (δ in ppm) of L1 - L4 (experiment results).
L1 |
L2 |
L3 |
L4 |
||||||
1H NMR |
13C NMR |
1H NMR |
13C NMR |
1H NMR |
13C NMR |
1H NMR |
13C NMR |
|
|
3.85 |
54.46 |
6.85 |
115.25 |
2.40 |
20.17 |
7.56 |
123.52 |
|
|
7.06 |
113.85 |
7.52 |
125.39 |
7.29 |
127.37 |
7.63 |
127.14 – 127.97 |
|
|
7.56 |
126.62 |
7.58 - 7.60 |
127.30 |
7.54 |
127.49 |
7.94 |
131.48 |
|
|
7.62 |
127.33 |
7.91 |
128.34 |
7.60 |
128.36 |
8.01 |
132.41 |
|
|
7.72 |
128.35 |
8.36 |
129.33 |
7.64 |
129.10 |
8.32 |
140.02 |
|
|
7.95 |
129.15 |
9.95 |
131.81 |
7.92 |
131.38 |
8.56 |
144.62 |
|
|
8.45 |
131.85 |
11.65 |
132.88 |
8.43 |
131.91 |
12.20 |
162.80 |
|
|
11.77 |
132.82 |
|
149.65 |
11.79 |
132.76 |
|
|
|
|
|
149.23 |
|
159.99 |
|
140.84 |
|
|
|
|
|
161.86 |
|
165.54 |
|
149.42 |
|
|
|
|
|
165.54 |
|
|
|
165.62 |
|
|
|
|
DFT calculations
The chemical reactivity descriptors of four aroyl hydrazone compounds (L1 - L4) were calculated using DFT methods based on Parr and Pearson’s framework and Koopman’s theorem [35]. Molecular orbital calculations predicted descriptors like Total energy (ETot), energy gap (ΔEGAP), dipole momentum (μ), electrophilicity (ω), and chemical hardness (η), which are detailed in Table 3.
Table 3 Global chemical reactivity descriptors for L1 - L4 calculated by the DFT method at B3LYP/6-31G*.
Quantum chemical descriptors |
L1 |
L2 |
L3 |
L4 |
ETot (eV) |
‒22.8694 |
‒21.7994 |
‒20.8221 |
‒25.3193 |
EHOMO (eV) |
‒5.86 |
‒5.69 |
‒5.89 |
‒6.61 |
ELUMO (eV) |
‒1.28 |
‒1.38 |
‒1.47 |
‒2.77 |
ΔEGAP (eV) |
4.58 |
4.31 |
4.42 |
3.83 |
η (eV) |
2.29 |
2.15 |
2.21 |
1.91 |
σ |
0.43 |
0.46 |
0.45 |
0.52 |
µ (Debye) |
1.65 |
0.93 |
1.87 |
1.10 |
χ (eV) |
3.57 |
3.53 |
3.68 |
4.69 |
ω |
2.78 |
2.90 |
3.06 |
5.74 |
Optimized molecular structures
As shown in Table 3, the total energy value calculated for the optimized geometry of each compound studied indicates that L4 has the lowest ETot value, suggesting that the conformation of L4 (Figure 5) is thermodynamically more stable than those of the L1 - L3. Moreover, the ΔEGAP value of L4 is also the smallest among the four compounds, implying that L4 facilitates the most efficient electron transfer between HUMO and LOMO orbitals [36,37]. The diminished HOMO-LUMO energy gaps reflect an enhancement in chemical reactivity, although it concurrently implies a reduction in chemical stability [38].
Frontier molecular orbitals (FMOs)
The electronic properties, including chemical stability and reactivity, of bioactive compounds can be largely deduced from their HOMO and LUMO orbital analysis and associated energy states. High molecular stability is directly correlated with a significant HOMO-LUMO energy difference, making this gap essential for determining the moleculeʼs stability index [39]. In general, a high EHOMO value is associated with a molecule’s capacity to donate electrons [40], while a lower ELUMO value reflects its ability to accept electrons [41]. As illustrated in Figure 5, density associated with the HOMO and LUMO orbitals are delocalized across the entire molecular structure of the molecules L1 - L4. In the visual representation, red solid surfaces denote the positive phase of each molecule, while green surfaces represent the negative phase [42]. The observed HOMO densities are primarily correlated to the strong electronegativity of oxygen and nitrogen atoms. According to the order of EHOMO listed in Table 3, the order is L2 (‒5.69) > L1 (‒5.86) > L3 (‒5.89) and L4 (‒6.61), suggesting that L2 has the greatest electron donating ability. This suggests a higher potential for antioxidant activity, as enhanced HOMO energies are often associated with enhanced radical scavenging activity. Conversely, L1 exhibited the lowest ELUMO value (−1.28), indicating its high electron acceptance potential. Dipole moment is another critical descriptor affecting molecular interactions and effectiveness within a biological system, particularly through hydrogen bonding, dipole-dipole interactions, and electrostatic interactions [43]. The order of dipole moment values follows the tren L3 > L1 > L4 > L2, indicating that L3 may interact more strongly with polar environments. Chemical hardness (η), which suggests a molecule’s resistance to charge transfer or polarization of the electron cloud during chemical reactions, is inversely related to molecular reactivity. Hard molecules generally exhibit a high energy gap, while soft molecules display a small energy gap [44]. Molecules with lower hardness and higher softness may have greater inhibition efficiency [45]. The hardness values of the molecules in this study decrease in the following order L1 > L3 > L2 > L4, suggesting that L4 is the softest and, consequently, the most chemically reactive and least stable compound among the four. The electrophilicity (ω), a measure of electron-accepting ability, was observed in the following descending order L4 > L3 > L2 > L1. Higher electrophilicity values generally suggest an enhanced ability to participate in electrophilic interactions, indicating that L4 possesses the most favorable electronic characteristics for such behavior. The potential of a compound as an ideal anticancer agent often requires a balance among its stability, reactivity, and interaction capability. Based on these three parameters, L1 and L3 emerge as the strongest candidates for anticancer compounds. L1 exhibits the high electron-donating potential, coupled with highest molecular stability and high interaction capability with polar environments. L3 possesses the highest capacity for interaction with polar molecular environments (due to its high dipole moment), along with high molecular stability and high electrophilicity.
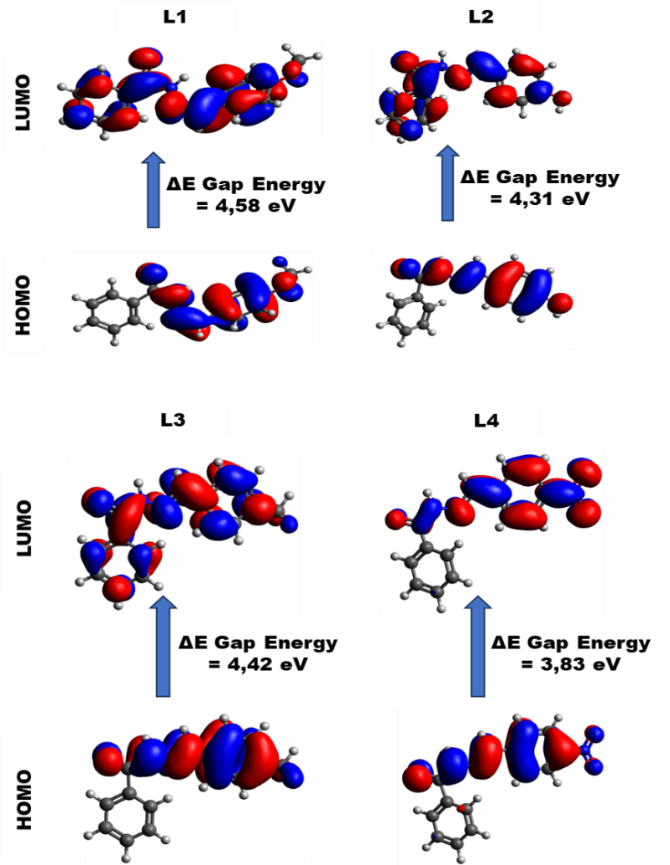
Figure 5 Optimized molecular structures and frontier molecular orbitals density distributions of L1 - L4.
Anticancer evaluation
Antioxidant activity
The IC50 values of all samples are presented in Table 4. The results indicated that all tested compounds demonstrated notable antioxidant activity, as evidenced by their low IC50 values. Among them, L2 showed the most potent antioxidant effect, with the lowest IC50 value of 0.19 ± 0.24 µg/mL and the highest % inhibition of 76.23 ± 3.97%. In addition, compounds L1, L2, and L3 exhibited superior antioxidant performance than the references. According to the findings, all synthesized aroylhydrazones derivatives exhibit potent antioxidant activity, as defined by IC50 values below 50 µg/mL [46].
Table 4 Antioxidant activity (IC50 and % inhibition) of the examined aroyl hydrazone derivatives.
Compound |
IC50 (µg/mL) |
% Inhibition |
Gallic Acid |
1.44 ± 0.15 |
70.95 ± 3.43 |
Quercetin |
1.69 ± 0.13 |
65.15 ± 2.28 |
L1 |
1.42 ± 1.28 |
53.02 ± 0.85 |
L2 |
0.19 ± 0.24 |
76.23 ± 3.97 |
L3 |
0.49 ± 0.36 |
56.86 ± 1.51 |
L4 |
13.30 ± 10.41 |
73.76 ± 1.70 |
Cytotoxicity
The cytotoxicity effects of the samples were tested against HeLa cell lines using the Presto blue method. The primary parameter applied to examine the cytotoxicity of the compounds was the IC50 value, with cisplatin serving as a positive control. The effect of varying concentrations of the aroylhydrazone derivatives on HeLa cell line viability after 48-hour incubation is depicted in Figure 6, while the corresponding IC50 values are presented in Table 5. All samples demonstrated anticancer activity against HeLa cells with IC₅₀ values ranging from 42 to 288 µg/mL. These values were notably higher than cisplatin, the positive control, indicating comparatively lower potency. Among the tested samples, L1 and L3 demonstrated moderate anticancer activity with IC50 values of 82.12 and 42.64 µg/mL, respectively, after 48 h of exposure. Meanwhile, with the same treatment, L2 and L4 exhibited weaker anticancer activity, with IC50 values of 287.40 and 213.50 µg/mL, respectively [47]. The variation in anticancer activity among the aroylhydrazone derivatives is due to structural differences, particularly the nature of the aromatic substituents at the para-position of the benzaldehyde moiety. These substituent effects influence the electronic characteristics and their possible interactions with biological targets within cells, thereby affecting their biological activity [48].
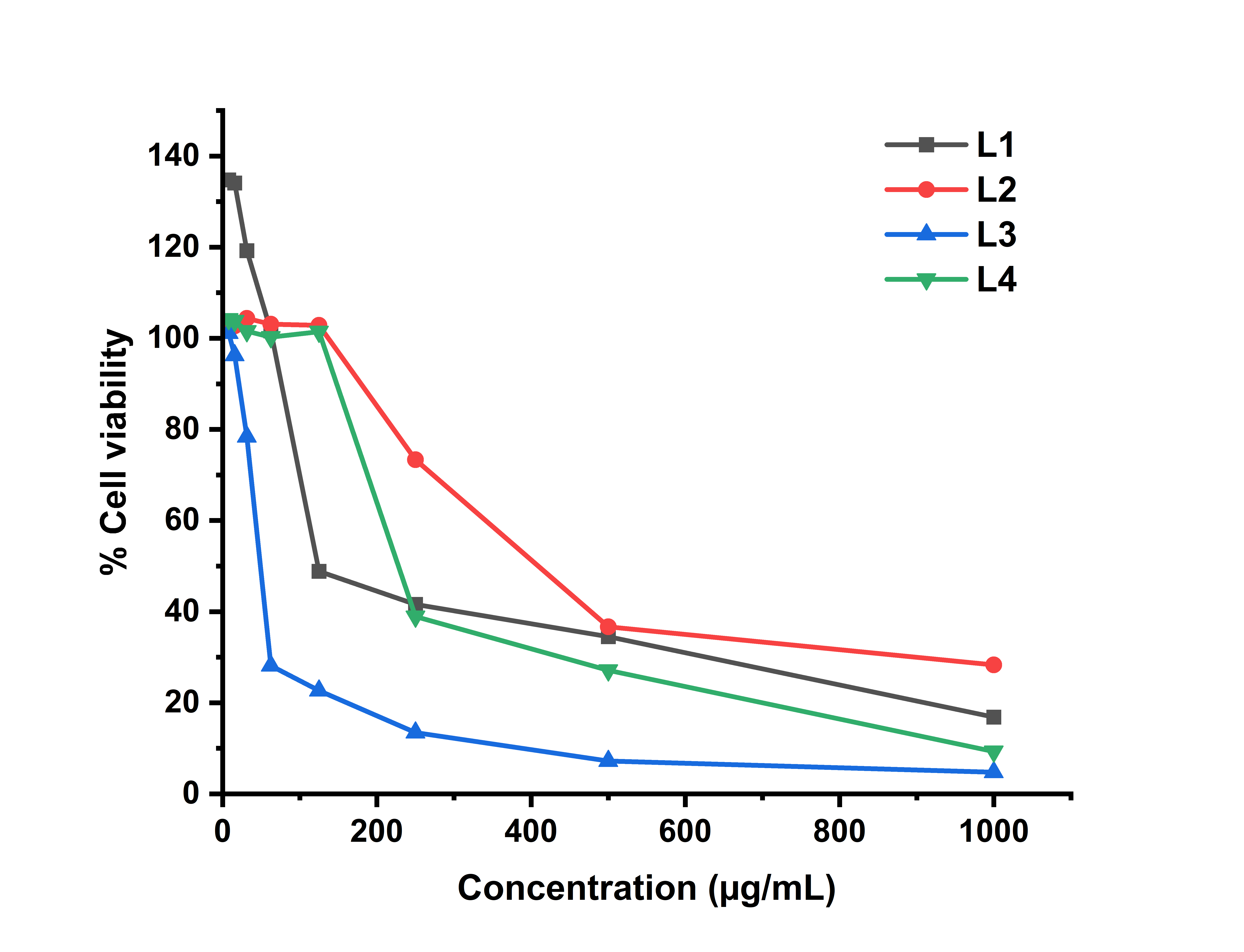
Figure 6 Cell viability vs aroylhydrazone concentration.
Table 5 Cytotoxic test of compounds against HeLa cells after 48 h of treatment.
Compounds |
IC50 |
(µg/mL) |
|
L1 |
82.12 |
L2 |
287.40 |
L3 |
42.64 |
L4 |
213.50 |
Cisplatin |
19.26 |
ADMET
The drug-likeness of the synthesized hydrazone derivatives was examined using Lipinski’s rule, which serves as a guideline to assess the oral bioavailability of potential drug candidates. According to this rule, compounds with oral activity generally exhibit a maximum of 5 hydrogen bond donors, 10 hydrogen bond acceptors, a molecular weight less than 500 Daltons, and a logD less than 5. A maximum of one violation is tolerable for a compound to be considered drug-like [49]. As shown in Table 6, all synthesized hydrazones met the criteria mentioned above without any violations, indicating favorable drug-likeness and solubility profiles. Moreover, the toxicity prediction results classified all compounds as relatively safe species, with no indications of mutagenicity, carcinogenicity, skin, or respiratory irritation. The yields indicate that the prepared compounds possess promising ADMET characteristics, highlighting their potential as viable candidates for further drug development.
Table 6 The values of the physicochemical and pharmacokinetic aspects (ADMET) of the synthesized compounds.
Compounds |
logD |
logS |
TPSA |
MW |
nHA |
nHD |
Lipinski rule |
Toxicity risk |
|||
Mut |
Car |
Irr |
Res |
||||||||
L4 |
3.08 |
‒4.67 |
84.60 |
269.08 |
6.0 |
1.0 |
0 |
no |
no |
no |
no |
L3 |
3.48 |
‒5.05 |
41.46 |
238.11 |
3.0 |
1.0 |
0 |
no |
no |
no |
no |
L2 |
2.53 |
‒3.84 |
61.69 |
240.09 |
4.0 |
2.0 |
0 |
no |
no |
no |
no |
L1 |
2.94 |
‒3.59 |
50.69 |
254.11 |
4.0 |
1.0 |
0 |
no |
no |
no |
no |
Erlotinib |
2.99 |
‒3.65 |
74.73 |
393.17 |
7.0 |
1.0 |
0 |
no |
no |
no |
no |
Doxorubicin |
3.07 |
‒4.67 |
84.60 |
269.08 |
6.0 |
1.0 |
0 |
no |
no |
no |
no |
*Drugs Reference: Topological polar surface area (TPSA); Molecular weight (MW); The logarithm of the n-octanol/water distribution coefficient (logD); The logarithm of aqueous solubility (logS); Number of hydrogen bond donors (nHD); Number of hydrogen bond acceptors (nHA); Lipinski’s Rule; (Mut) mutagenic; (Car) Carcinogenicity; (Irr) irritant; (Res) Respiratory.
Molecular docking analysis
Molecular docking simulations were performed to analyze the binding interactions and associated energies between the synthesized ligands and the active sites of Cyclin-Dependent Kinase 2(CDK2). These simulations help in elucidating the nature and strength of molecular interactions, providing insights into the inhibitory potential of the compounds. CDK2 is a widely recognized target in cancer therapy research because of its essential function in regulating the cell cycle [50]. The binding energies and estimated inhibition constants for each ligand-enzyme complex are summarized in Table 8. The detailed molecular interaction profiles between CDK2 and the ligands L1 - L4 are visually depicted in Figure 7. Overall, hydrogen bonding is crucial in modulating the interaction between inhibitors and their target receptors. The molecular interaction diagrams reveal various interactions that contribute to the stability of the ligand-receptor complexes, as summarized below:
According to Table 7, ligand L1 involves in several interactions, including a Pi-sigma bond with Phe80 (3.78 Å), involving the hydrogen atom of the methoxy group, and multiple Pi-alkyl interactions with Ile10; Ala31; Val64; Ala144; Leu134 (5.01;3.66;5.05;4.94;4.55 Å) through its aromatic ring. L1 exhibits an unfavorable electrostatic (positive-positive) interaction with His84 (4.99 Å), indicating possible repulsion between positively charged moieties. Ligand L2 forms a conventional hydrogen bond with Glu81 (2.29 Å) through the nitrogen of the amine group in the hydrazone. It also establishes Pi-sigma bonds with Leu134 (3.91 Å), Pi-cation bonds with His84 (5.00 Å), Pi-alkyl bonds with Ile10, Val18, Ala31, Val64 (4.62;4.93;5.05;5.39 Å), and T-shaped Pi-Pi interactions with Phe80 (5.67 Å) involving benzene groups. Ligand L3 forms a hydrogen bond with Glu81 (2.35 Å) via the hydrazone amine nitrogen. In addition, there are Pi-alkyl bonds with Ile10; Ala31; Val64 (4.53;5.28;5.13 Å) via benzene, Pi-sigma bond with Leu134 (3.79 Å) interacting with the benzene group, and a T-shaped Pi-Pi bond with Phe80 (5.69 Å) via benzene. Meanwhile, ligand L4 forms a hydrogen bond with Glu81 (2.34 Å), which involves nitrogen interaction of the amine group in hydrazone. In addition, there are Pi-alkyl bonds with Ile10; Ala31; Val64; and Val18 (4.57;5.07;5.38;4.98 Å) via benzene, and a Pi-sigma bond with Leu134 and Ala144 (3.79 and 3.85 Å) which interact with the benzene group, hydrogen bond with Gln85 (5.69 Å) via the oxygen of the nitro group. In the Roniciclib standard, two hydrogen bonds are formed, namely with ASN132 (3.01 Å) and ASP36 (1.77 Å), which occur through the interaction of oxygen atoms bound to phenol and sulfonyl groups. In addition, there are Pi-Alkyl interactions with Phe80, AIA144, VAL64, ALA31, and ILE10 (4.13;4.27;4.40;4.36;4.52 Å) involving fluorine atoms and benzene rings. In addition, Pi-sigma interactions occur with Leu134 (3.16 Å), which involve benzene groups. Hydrophobic interactions such as Pi-cation, Pi-Pi T-shaped, and Pi-alkyl are primarily driven by van der Waals forces. In contrast, hydrogen bonding and electrostatic interactions contribute to the hydrophilic profile of the ligand-receptor complexes [51]. Notably, all synthesized compounds L1 - L4 display Pi-sigma interactions, which are absent in the binding profile of Roniciclib. This distinct interaction pattern may suggest enhanced binding affinity and improved inhibitory properties. These observations further imply that nucleophilic regions of the receptor, particularly those involving hydroxyl and amine functional groups, play a significant role in mediating strong ligand–amino acid interactions.
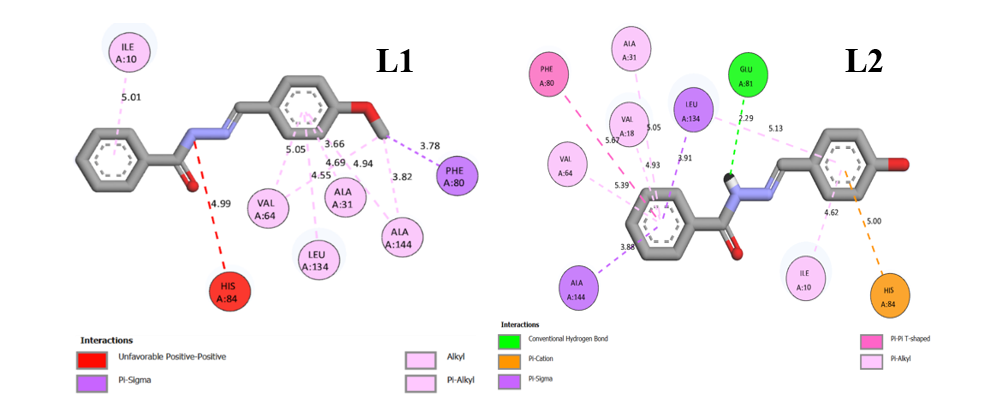
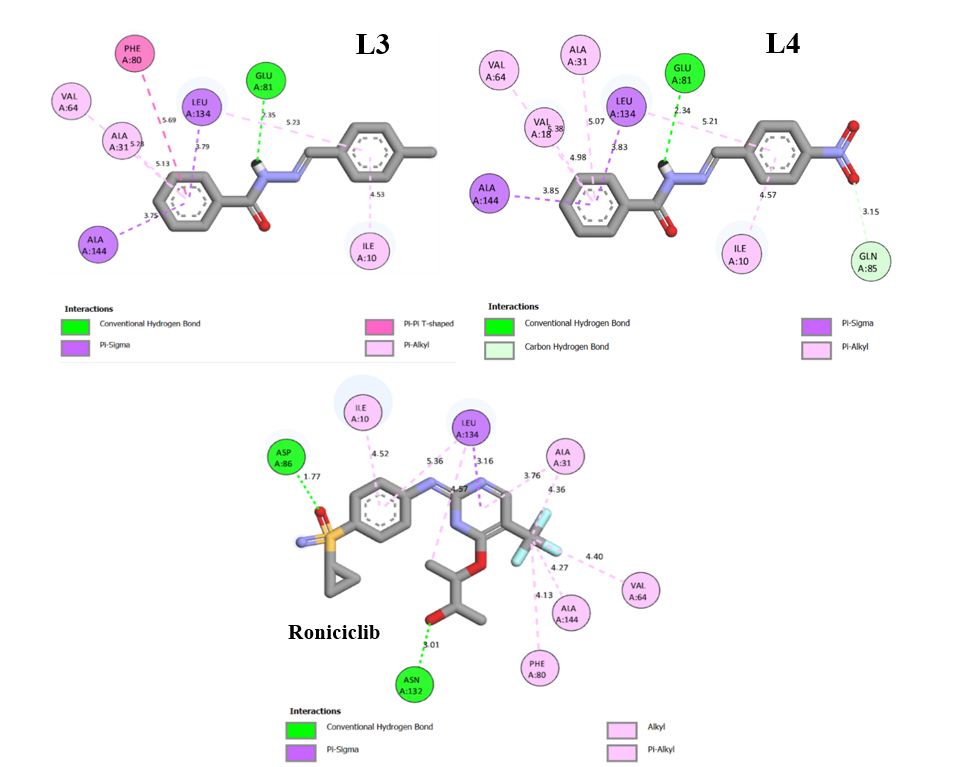
Figure 7 Binding-interaction diagrams of L1, L2, L3, L4 and Roniciclib with CDK2.
Table 7 Molecular docking results.
Compounds |
Amino acid interactions |
Distance (Å) |
Type of interaction |
L1 |
- HIS84 - PHE80 - ILE10, ALA31, VAL64, ALA144, LEU134, VAL64, ALA144 |
4.99 3.78 5.01, 3.66, 5.05, 4.94, 4.55 4.69, 3.82 |
Unfavorable positive-positive Pi-sigma Pi-alkyl
|
L2 |
- GLU81 - HIS84 - LEU134 - PHE80 -ILE10, VAL18, ALA31, VAL64 |
2.29 5.00 3.91 5.67 4.62, 4.93, 5.05, 5.39 |
Hydrogen bond Pi-cation Pi-sigma Pi-Pi T-shaped Pi-alkyl |
L3 |
- GLU81 - LEU134 - PHE80 -ILE10, ALA31, VAL64 |
2.35 3.79 5.69 4.53, 5.28, 5.13 |
Hydrogen bond Pi-sigma Pi-Pi T-shaped Pi-alkyl |
L4 |
- GLU81 - GLN85 -LEU134, ALA144 -ILE10, VAL18, ALA31, VAL64 |
2.34 1.35 3.83, 3.85 4.57, 4.98, 5.07, 5.38 |
Hydrogen bond Carbon hydrogen bond Pi-sigma Pi-alkyl |
Roniciclib |
-ASN132, ASP86 - LEU134 -PHE80, ALA144, VAL64, ALA31, ILE10 |
3.01, 1.77 3.16 4.13, 4.27, 4.40, 4.36, 4.52 |
Hydrogen bond Pi-sigma Pi-alkyl |
Table 8 compares the binding energies and inhibition constants (Ki) of compounds studied, showing Roniciclib > L4 > L1 > L2 > L3 in binding affinity. Ligand L4 has the best docking score and inhibition constant (Ki) of 17.80 nM, with a binding energy of −10.57 kcal/mol, indicating strong binding affinity. These findings suggest L4 potential as a CDK2 inhibitor, akin to the native reference compound, making it a promising candidate for CDK2-targeted therapies.
Table 8 Comparison of the binding energy and inhibition constant (Ki) of all samples and Roniciclib.
Compounds |
Ki (nM) |
Binding energy (kcal/mol) |
L1 |
136.00 |
‒9.37 |
L2 |
136.70 |
‒9.36 |
L3 |
210.05 |
‒9.11 |
L4 |
17.80 |
‒10.57 |
Roniciclib |
1.20 |
‒12.17 |
Conclusions
Four aroyl hydrazone derivative compounds were successfully synthesized and characterized. The structure compounds were comprehensively investigated using UV-Vis, FTIR, 1H NMR, and 13C NMR techniques. Quantum chemical analysis was done with the DFT method employing the 6-31G* basis set, and the theoretical FTIR and UV-Vis spectra showed good correlation with the experimental data. The calculated ΔEGAP values indicated that L1 and L3 possessed the highest molecular stability and high chemical reactivity. In vitro, antioxidant evaluation based on the DPPH assay exhibited that L1 - L3 had superior antioxidant activities compared to the standard antioxidants. Cytotoxicity studies against HeLa cells indicated that all four compounds exhibited anticancer activity, with L1 and L3 showing moderate potency. In addition, the predicted ADMET and pharmacokinetic aspects revealed that these four aroylhydrazone compounds possess favorable oral bioavailability, low toxicity, and promising drug-likeness profiles. Molecular docking simulations supported these findings, revealing strong binding affinities of the ligands to the CDK2 protein. Notably, L4 exhibited the lowest binding energy and inhibition constant, comparable to the reference inhibitor Roniciclib, suggesting its potential as a CDK2 inhibitor involved in cancer cell proliferation. Overall, the synthesized aroylhydrazone derivatives demonstrate significant antioxidant and anticancer properties, supported by both experimental and computational data. These compounds represent promising candidates for further development as novel therapeutic agents targeting oxidative stress and cervical cancer.
Acknowledgements
The authors convey their heartfelt appreciation to the Chancellor of the Sepuluh Nopember Institute of Technology (ITS) for providing the essential research infrastructure. The authors also thank the assistance granted through the Indonesian Education Scholarship Program (Beasiswa Pendidikan Indonesia, BPI) managed by the Center for Higher Education Funding and Assesment (PPAPT), operating under the Ministry of Higher Education, Science, and Technology of Republic Indonesia (Kemendikti Saintek RI). Further appreciation is directed to the Indonesia Endowment Fund for Education (LPDP) under the Ministry of Finance of Republic Indonesia for the granted fellowship.
Declaration of Generative AI in Scientific Writing
The authors acknowledge the use of generative AI (specifically Gemini by Google) during the preparation of this manuscript for language editing and grammatical corrections. No content generation or data interpretation was performed by the AI. The authors remain fully responsible for the content and conclusions of this work.
CRediT Author Statement
Imanuel Gauru: Conceptualization, Writing – original draft, Methodology, Visualization, Validation, Software, Validation. Yusuf Syahril Alam: Supervision, Software, Resources, Conceptualization. Syafri Izzat Abidiy: Visualization, Validation, Software. Fidelis Nitti: Writing – review & editing, Formal analysis. Holilah Holilah: Formal analysis, Data curation, Validation. Yuly Kusumawati: Visualization, Validation, Software, Writing – review & editing. Mardi Santoso: Writing – review & editing, Formal analysis. Arif Fadlan: Writing – review & editing, Validation, Formal analysis. Fahimah Martak: Conceptualization, Writing – review & editing, Supervision, Validation, Resources.
References
[1] J Pisk, I. Đilović, T Hrenar, D Cvijanović, G Pavlović and V Vrdoljak. Effective methods for the synthesis of hydrazones, quinazolines, and Schiff bases: Reaction monitoring using a chemometric approach. RSC Advances 2020; 10(63), 38566-38577.
[2] C Arunagiri, AG Anitha, A Subashini and S Selvakumar. Synthesis, X-ray crystal structure, vibrational spectroscopy, DFT calculations, electronic properties and Hirshfeld analysis of (E) -4-Bromo-N’-(2,4-dihydroxy-benzylidene) benzohydrazide. Journal of Molecular Structure 2018; 1163, 368-378.
[3] VFS Pape, S Tóth, A Füredi, K Szebényi, A Lovrics, P Szabó, M Wiese and G Szakács. Design, synthesis and biological evaluation of thiosemicarbazones, hydrazinobenzothiazoles and arylhydrazones as anticancer agents with a potential to overcome multidrug resistance. European Journal of Medicinal Chemistry 2016; 117, 335-354.
[4] A Kajal, S Bala, N Sharma, S Kamboj and V Saini. Therapeutic potential of hydrazones as anti-inflammatory agents. International Journal of Medicinal Chemistry 2014; 2014, 761030.
[5] SG Küçükgüzel, A Mazi, F Sahin, S Öztürk and J Stables. Synthesis and biological activities of diflunisal hydrazide-hydrazones. European Journal of Medicinal Chemistry 2003; 38(11-12), 1005-1013.
[6] JV Ragavendran, D Sriram, SK Patel, IV Reddy, N Bharathwajan, J Stables and P Yogeeswari. Design and synthesis of anticonvulsants from a combined phthalimide-GABA-anilide and hydrazone pharmacophore. European Journal of Medicinal Chemistry 2007; 42(2), 146-151.
[7] MA Abdelrahman, I Salama, MS Gomaa, MM Elaasser, MM Abdel-Aziz and DH Soliman. Design, synthesis and 2D QSAR study of novel pyridine and quinolone hydrazone derivatives as potential antimicrobial and antitubercular agents. European Journal of Medicinal Chemistry 2017; 138, 698-714.
[8] S Vogel, D Kaufmann, M Pojarová, C Müller, T Pfaller, S Kühne, PJ Bednarski and EV Angerer. Aroyl hydrazones of 2-phenylindole-3-carbaldehydes as novel antimitotic agents. Bioorganic & Medicinal Chemistry 2008; 16(12), 6436-6447.
[9] C Vanucci-Bacqué, C Carayon, C Bernis, C Camare, A Nègre-Salvayre and F Bedos-Belval. Synthesis, antioxidant and cytoprotective evaluation of potential antiatherogenic phenolic hydrazones. A structure-activity relationship insight. Bioorganic & Medicinal Chemistry 2014; 22(15), 4269-4276.
[10] Jasril, Y Nurulita, N Afriana, I Ikhtiarudin and N Frimayanti. Synthesis, docking, and molecular dynamic study of hydrazones compounds to search potential inhibitor for breast cancer MCF-7. Thai Journal of Pharmaceutical Sciences 2021; 45(6), 477-486.
[11] V Balachandran, G Santhi, V Karpagam and A Lakshmi. DFT computation and spectroscopic analysis of N-(p-methoxybenzylidene) aniline, a potentially useful NLO material. Journal of Molecular Structure 2013; 1047, 249-261.
[12] Naveen, RK Tittal, VD Ghule, N Kumar, L Kumar and K Lal. Design, synthesis, biological activity, molecular docking and computational studies on novel 1,4-disubstituted-1,2,3-Triazole-Thiosemicarbazone hybrid molecules. Journal of Molecular Structure 2020; 1209, 127951.
[13] C James, AA Raj, R Reghunathan, VS Jayakumar and IH Joe. Structural conformation and vibrational spectroscopic studies of 2,6-bis(p-N,N-dimethyl benzylidene) cyclohexanone using density functional theory. Journal of Raman Spectroscopy 2006; 37(12), 1381-1392.
[14] Nurlelasari, I Rahmayanti, S Salam, A Safari, D Harneti, R Maharani, AT Hidayat, M Tanjung, R Retnowati, Y Shiono and U Supratman. A new havanensin-type limonoid from Chisocheton macrophyllus. Applied Biological Chemistry 2021; 64, 35.
[15] C Thepmalee, N Sawasdee, S Thongyim, N Poungvarin, PT Yenchitsomanus and A Panya. Enhancing T cell cytotoxicity against lung cancer with an αPD-L1 protein engager and gemcitabine combination therapy. Biomedicine & Pharmacotherapy 2025; 188, 118161.
[16] J Branković, N Milivojevic, V Milovanovic, D Simijonovic, ZD Petrovic and Z Markovic. Evaluation of antioxidant and cytotoxic properties of phenolic N-acylhydrazones: Structure-activity relationship. Royal Society Open Science 2022; 9(6), 211853.
[17] YP Pasaribu, A Fadlan, S Fatmawati and T Ersam. Biological activity evaluation and in silico studies of polyprenylated benzophenones from Garcinia celebica. Biomedicines 2021; 9(11), 1654.
[18] A Ragab, YA Ammar, A Ezzat, AM Mahmoud, MBI Mohamed, AS El-Tabl and RS Farag. Synthesis, characterization, thermal properties, antimicrobial evaluation, ADMET study, and molecular docking simulation of new mono Cu (II) and Zn (II) complexes with 2-oxoindole derivatives. Computers in Biology and Medicine 2022; 145, 105473.
[19] SA Rupa, MR Moni, MAM Patwary, MM Mahmud, MA Haque, J Uddin and SMT Abedin. Synthesis of novel tritopic hydrazone ligands: Spectroscopy, biological activity, DFT, and molecular docking studies. Molecules 2022; 27(5), 1656.
[20] L Adjissi, N Chafai, K Benbouguerra, I Kirouani, A Hellal, H Layaida, M Elkolli, C Bensouici and S Chafaa. Synthesis, characterization, DFT, antioxidant, antibacterial, pharmacokinetics and inhibition of SARS-CoV-2 main protease of some heterocyclic hydrazones. Journal of Molecular Structure 2022; 1270, 134005.
[21] YS Alam, P Pudjiastuti, S Malulana, NR Affifah, F Martak and A Fadlan. Synthesis and antidiabetic evaluation of N’-Benzylidenebenzohydrazide derivatives by in silico studies. Indonesian Journal of Chemistry 2023; 23(4), 1061-1070.
[22] PC Agu, CA Afiukwa, OU Orji, EM Ezeh, IH Ofoke, CO Ogbu, EI Ugwuja and PM Aja . Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Scientific Reports 2023; 13, 13398.
[23] I Al-Qadsy, AB Al-Odayni, WS Saeed, A Alrabie, A Al-Adhreai, LAS Al-Faqeeh, P Lama, AA Alghamdi and M Farooqui. Synthesis, characterization, single-crystal x-ray structure and biological activities of [(Z)-N -(4-Methoxybenzylidene)benzohydrazide-Nickel(II)] complex. Crystals 2021; 11(2), 110.
[24] W Yu, G Huang,Y Zhang, H Liu, L Dong, X Yu, Y Li and J Chang. I2-mediated oxidative C−O bond formation for the synthesis of 1,3,4-Oxadiazoles from aldehydes and hydrazides. The Journal of Organic Chemistry 2013; 78(20), 10337-10343.
[25] S Nagar, S Raizada and N Tripathee. A review on various green methods for synthesis of Schiff base ligands and their metal complexes. Results in Chemistry 2023; 6, 101153.
[26] J Silver. Let us teach proper thin layer chromatography technique. Journal of Chemical Education 2020; 97(12), 4217-4219.
[27] K Benbouguerra, N Chafai, S Chafaa, YI Touahria and H Tlidjane. New α-Hydrazinophosphonic acid: Synthesis, characterization, DFT study and in silico prediction of its potential inhibition of SARS-CoV-2 main protease. Journal of Molecular Structure 2021; 1239, 130480.
[28] N Chafai, S Chafaa, K Benbouguerra, A Hellal and M Mehri. Synthesis, spectral analysis, anti-corrosive activity and theoretical study of an aromatic hydrazone derivative. Journal of Molecular Structure 2019; 1181, 83-92.
[29] NA Mathews, A Jose and MRP Kurup. Synthesis and characterization of a new aroylhydrazone ligand and its cobalt(III) complexes: X-ray crystallography and in vitro evaluation of antibacterial and antifungal activities. Journal of Molecular Structure 2019; 1178, 544-553.
[30] K Ahmed, M Bashir, R Bano, M Sarfraz, HU Khan, S Khan, A Sharif, A Waseem, MA Gilani, K Batool, R Idrees, A Rauf, RSZ Saleem and M Arshad. Potent heteroaromatic hydrazone based 1,2,4-triazine motifs: Synthesis, anti-oxidant activity, cholinesterase inhibition, quantum chemical and molecular docking studies. Journal of Molecular Structure 2023; 1284, 135383.
[31] P Teli, S Soni, S Teli and S Agarwal. Unveiling the catalytic potency of a novel hydrazone-linked covalent organic framework for the highly efficient one-pot synthesis of 1,2,4-triazolidine-3-thiones. Nanoscale Advances 2024; 6(22), 5568-5578.
[32] S Ibesh, Y Bitar and S Trefi. A New method for simultaneous qualitative and quantitative determination of amlodipine besylate and atorvastatin calcium in bulk and pharmaceutical formulations using transmission FT-IR spectroscopy. Heliyon 2023; 9(3), 14189.
[33] EH El-Mossalamy, NF Al-Harby, SA Aal, NM Ali, M El-Desawy, MM Elewa and ME Batouti. Tenability on schiff base Hydrazone derivatives and Frontier molecular orbital. Heliyon 2024, 10(2), 24472.
[34] R Munir, N Javid, M Zia-Ur-Rehman, M Zaheer, R Huma, A Roohi and MM Athar. Synthesis of novel N-acylhydrazones and their C-N/N-N bond conformational characterization by NMR spectroscopy. Molecules 2021; 26(16), 4908.
[35] N Flores-Holguín, J Frau and D Glossman-Mitnik. In silico pharmacokinetics, ADMET study and conceptual DFT analysis of two plant cyclopeptides isolated from rosaceae as a computational peptidology approach. Frontiers in Chemistry 2021; 9, 708364.
[36] NM Sabry, R Badry, FK Abdel-Gawad, H Elhaes and MA Ibrahim. Electronic structure, global reactivity descriptors and nonlinear optical properties of glycine interacted with ZnO, MgO and CaO for bacterial detection. Scientific Reports 2024; 14, 22801.
[37] R Morad, M Akbari and M Maaza. Theoretical study of chemical reactivity descriptors of some repurposed drugs for COVID-19. MRS Advances 2023; 8, 656-660.
[38] MA Mumit, TK Pal, MA Alam, MAAAA Islam, S Paul and MC Sheikh. DFT studies on vibrational and electronic spectra, HOMO-LUMO, MEP, HOMA, NBO and molecular docking analysis of benzyl-3-N-(2,4,5-trimethoxyphenylmethylene)hydrazinecarbodithioate. Journal of Molecular Structure 2020; 1220, 128715.
[39] YT Male, IW Sutapa, IB Kapelle and M Lopulalan. QSAR modeling and design of a new model of anti- HIV drug 1-aryl-tetrahydroisoquinoline derived using the PM3 semiempirical method. RASAYAN Journal of Chemistry 2022; 15(1), 359-368.
[40] HA Arjun, GNA Kumar, R Elancheran and S Kabilan. Crystal structure, DFT and Hirshfeld surface analysis of (E)-N′-[(1-chloro-3,4-dihydronaphthalen-2-yl)methylidene]benzohydrazide monohydrate. Acta Crystallographica Section E Crystallographic Communications 2020; 76(2), 132-136.
[41] U Vanitha, R Elancheran, V Manikandan, S Kabilan and K Krishnasamy. Design, synthesis, characterization, molecular docking and computational studies of 3-phenyl-2-thioxoimidazolidin-4-one derivatives. Journal of Molecular Structure 2021; 1246, 131212.
[42] N Chafai, S Chafaa, K Benbouguerra, D Daoud, A Hellal and M Mehri. Synthesis, characterization and the inhibition activity of a new α-aminophosphonic derivative on the corrosion of XC48 carbon steel in 0.5 M H2SO4: Experimental and theoretical studies. Journal of the Taiwan Institute of Chemical Engineers 2017; 70, 331-344.
[43] H Kang and SG Lee. Scalable analysis of dipole moment fluctuations for characterizing intermolecular interactions and structural stability. Journal of Chemical Information and Modeling 2024; 64(11), 4518-4529.
[44] TK Chaitra, KN Mohana, DM Gurudatt and HC Tandon. Inhibition activity of new thiazole hydrazones towards mild steel corrosion in acid media by thermodynamic, electrochemical and quantum chemical methods. Journal of the Taiwan Institute of Chemical Engineers 2016; 67, 521-531.
[45] M Rafiq, M Khalid, MN Tahir, MU Ahmad, MU Khan, MM Naseer, AAC Braga, S Muhammad and Z Shafiq. Synthesis, XRD, spectral (IR, UV-Vis, NMR) characterization and quantum chemical exploration of benzoimidazole-based hydrazones: A synergistic experimental-computational analysis. Applied Organometallic Chemistry 2019; 33(11), 5182.
[46] A Itam, MS Wati, V Agustin, N Sabri, RA Jumanah and M Efdi. Comparative study of phytochemical, antioxidant, and cytotoxic activities and phenolic content of Syzygium aqueum (Burm. f. Alston f.) extracts growing in West Sumatera Indonesia. The Scientific World Journal 2021; 2021, 5537597.
[47] GU Anywar, E Kakudidi, H Oryem-Origa, A Schubert and C Jassoy. Cytotoxicity of medicinal plant species used by traditional healers in treating people suffering from HIV/AIDS in uganda. Frontiers in Toxicology 2022; 4, 832780.
[48] Mİ Han and N İmamoğlu. Design, synthesis, and anticancer evaluation of novel tetracaine hydrazide-hydrazones. ACS Omega 2023; 8(10), 9198-9211.
[49] L Fu , S Shi , J Yi , N Wang , Y He , Z Wu , J Peng , Y Deng , W Wang , C Wu , A Lyu , X Zeng , W Zhao , T Hou and D Cao. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Research 2024; 52(1), 422-431.
[50] J Zhang, Y Gan, H Li, J Yin, X He, L Lin, S Xu, Z Fang, B Kim, L Gao, L Ding, E Zhang, X Ma, J Li, L Li, Y Xu, D Horne, R Xu, H Yu, Y Gu and W Huang. Inhibition of the CDK2 and cyclin a complex leads to autophagic degradation of CDK2 in cancer cells. Nature Communications 2022; 13, 2835.
[51] T Topal, Y Zorlu and N Karapınar. Synthesis, X-ray crystal structure, IR and Raman spectroscopic analysis, quantum chemical computational and molecular docking studies on hydrazone-pyridine compound: As an insight into the inhibitor capacity of main protease of SARS-CoV2. Journal of Molecular Structure 2021; 1239, 130514.