Trends
Sci.
2026;
23(8):
13450
Temporal Immune Trajectory and Candidate Biomarkers in Sepsis Associated with Community-Acquired Pneumonia: A Systematic Review
Suparto1,2, Suwarman3, Wani Devita Gunardi4, Henny Tannady Tan5,
Nathalie Widjaja2 and Nur Atik6,*
1Doctoral Study Program, Faculty of Medicine, Universitas Padjadjaran, West Java, Indonesia.
2Department of Anesthesiology, Faculty of Medicine and Health Sciences, Universitas Kristen Krida Wacana,
Jakarta, Indonesia
3Department of Anesthesiology and Intensive Care, Universitas Padjajaran, West Java, Indonesia
4Department of Microbiology, Faculty of Medicine & Health Sciences, Universitas Kristen Krida Wacana,
Jakarta, Indonesia
5Department of Internal Medicine, Faculty of Medicine and Health Sciences, Universitas Kristen Krida Wacana, Jakarta, Indonesia
6Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, West Java, Indonesia
(*Corresponding author’s e-mail: [email protected])
Received: 26 January 2026, Revised: 19 March 2026, Accepted: 29 March 2026, Published: 10 April 2026
Abstract
Community-acquired pneumonia (CAP) with sepsis remains a leading cause of morbidity and mortality worldwide. Although numerous immune biomarkers have been proposed, integrated evidence describing how immune responses evolve in CAP-associated sepsis is limited, particularly from blood transcriptomic studies. This systematic review aimed to synthesize evidence on temporal immune trajectories and identify candidate transcriptomic biomarkers associated with outcomes in CAP-associated sepsis. A systematic search of medRxiv, PubMed, and CENTRAL was conducted for studies reporting serial blood transcriptomic or immune biomarker data in adult patients with CAP-associated sepsis. Studies with clearly defined timepoints and clinical outcomes were included. Risk of bias was assessed using an adapted Newcastle-Ottawa Scale with transcriptomic-specific criteria. Due to heterogeneity in study design and timing definitions, findings were integrated using structured narrative synthesis. Six studies met the inclusion criteria. Early sepsis was characterized by consistent upregulation of innate inflammatory pathways, including neutrophil activation, interferon-related genes, and cytokine signaling, largely independent of pathogen type. Survivors demonstrated recovery of antigen presentation and adaptive immune pathways at later time points, whereas non-survivors exhibited persistent inflammatory signatures, impaired lymphocyte-associated transcription, immune checkpoint activation, and endothelial dysfunction. Recurrent candidate biomarkers included inflammatory gene signatures, immune endotype classifications, and transcriptomic indicators of adaptive immune suppression. CAP-associated sepsis follows dynamic immune trajectories rather than a single inflammatory state. Temporal transcriptomic patterns may help identify patients at risk of immune failure and guide immunomodulatory strategies. However, heterogeneity in timing definitions, small sample sizes, and limited validation underscore the need for prospective studies before clinical implementation.
Keywords: Community-acquired pneumonia, Sepsis, Transcriptomic, Biomarker
Introduction
Community-acquired pneumonia (CAP) is an infectious disease that causes global illness with a high risk of mortality [1]. This infection targets the lungs, with symptoms including fever, cough, and dyspnea. The severe condition of CAP may progress to acute respiratory distress syndrome (ARDS), pleural effusion, sepsis, and multi-organ failure. Furthermore, this condition affects vulnerable people, including the elderly, immunocompromised patients, and people with chronic comorbidities [2-4].
The spread of CAP depends on the pathogens involved, such as bacteria or viruses. Common bacteria included Streptococcus pneumoniae, Haemophilus influenzae, and Klebsiella pneumoniae, as well as atypical pathogens such as Mycoplasma pneumoniae and Legionella species [4]. Furthermore, influenza viruses and coronaviruses are prevalent in viral etiologies [5-7]. CAP with sepsis, which often occurs under severe conditions, has become a worldwide public health issue due to weakened immune systems. This condition represents a dysregulated host response to infection that leads to life-threatening organ dysfunction. Furthermore, the number of sepsis incidents has gradually increased in recent years, driven by more extended hospital stays, the rise of antibiotic resistance, the increased prevalence of invasive medical procedures, and advances in diagnostic technology [8,9]. This condition significantly affects patient prognosis. In addition, sepsis is not only the result of an excessive inflammatory response but also a complex and dynamic interaction between pro-inflammatory and immunosuppressive pathways that evolve throughout the course of disease [10].
During the early phases of sepsis, innate immune responses are often strongly activated [11]. This includes the secretion of cytokines and chemokines, mobilization of neutrophils, and activation of monocytes. Increasing evidence indicates that prolonged or persistent immune suppression, such as immunoparalysis, significantly contributes to adverse outcomes. Elevated levels of both pro-inflammatory and anti-inflammatory mediators have been associated with increased mortality rates [12]. These findings suggest that the trajectory and balance of immune responses over time, rather than the magnitude of inflammation alone, determine disease progression and clinical outcomes.
The management of sepsis from CAP often involves administering antibiotics to eliminate the source of infection, restoring hemodynamics, and supporting organ function. Alternative therapies that have been investigated include corticosteroids and immunosuppressive drugs, but their clinical benefits remain inconsistent across studies [3]. This suggested that the patients have varied responses. Recently, there has been increased focus on improving the immune system in the body.
Certain approaches involve altering immunological checkpoints and using cell-based treatments, including mesenchymal stem cells, which have been shown in earlier studies to modulate immune responses, lower inflammation, and promote tissue repair [13]. However, these strategies have been limited to use in actual clinical environments due to the lack of reliable sources and the inability to identify which patients would benefit most. Therefore, a deeper understanding of immune response dynamics in CAP-associated sepsis is needed to guide precision medicine approaches.
One important technique for understanding how the host immune system responds to sepsis and other serious illnesses is high-throughput whole-blood transcriptomic profiling. According to numerous studies of gene expression, a significant fraction of the human transcriptome is regulated differently during sepsis compared with healthy individuals or those without sepsis [14-16]. Pathways controlling T-cell function, hemostasis, cellular metabolism, antigen presentation, interferon signaling, and innate immunity are altered [17]. Regardless of the source of infection, transcriptomic analysis has facilitated the identification of genetic endotypes, or sepsis response signatures (SRS), that classify patients by their immunological status and clinical outcomes.
Recent studies have shown the importance of gene expression timing in understanding the immunopathology of sepsis, suggesting that sepsis is better characterized as a spectrum of immunological states rather than a singular disease [16]. Despite the growing availability of transcriptomic datasets, integrated synthesis of temporal immune responses in CAP-associated sepsis remains limited. Most existing studies focus on single time-point measurements or heterogeneous sepsis populations without distinguishing CAP as a specific clinical context. Consequently, the relationships among early inflammation, interferon responses, adaptive immune failure, immunological checkpoint engagement, and clinical outcomes have not been extensively integrated across the studies. In this context, the present systematic review addresses the following clinical question: Among adults with CAP-associated sepsis, what temporal transcriptomic patterns characterize immune responses over the course of disease, and which gene signatures are associated with survival or adverse outcomes?
The goal of this systematic review is to synthesize transcriptomic studies involving critically ill adult patients with CAP-associated sepsis. Specifically, this review aims to identify conserved gene signatures, immune pathways, and temporal immune trajectories associated with disease severity and mortality. Unlike other transcriptomic review studies that focus solely on static immune genes, this systematic review emphasizes the dynamic evolution of immune responses, including early innate hyperinflammation, interferon signaling, adaptive immune dysfunction, and immune checkpoint activation across bacterial and viral CAP-associated sepsis. By integrating findings across diverse cohorts and infection types, this study seeks to identify common molecular mechanisms underlying sepsis immunopathology and to highlight candidate potential biomarkers that may inform future precision immunomodulatory therapy.
Although immune dysregulation is a hallmark of sepsis, most prior studies have focused on single time-point biomarkers or heterogeneous sepsis populations without distinguishing CAP as a specific clinical entity. Moreover, while transcriptomic profiling has identified molecular endotypes in sepsis, integrated evidence describing temporal immune trajectories in CAP-associated sepsis remains limited. In particular, it is unclear which transcriptional patterns differentiate early hyperinflammation from later adaptive immune failure and how these trajectories relate to mortality. Furthermore, a structured synthesis of longitudinal blood transcriptomic data is warranted to better understand immune dynamics and identify clinically relevant biomarkers in CAP-associated sepsis. Therefore, this systematic review aimed to synthesize temporal blood transcriptomic studies in CAP-associated sepsis to identify conserved immune trajectories and candidate biomarkers associated with clinical outcomes.
Materials and methods
Study design and reporting standards
This systematic review of transcriptomic investigation examining host immune responses in sepsis was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 guidelines. The review protocol was registered on the Open Science Framework (OSF) prior to data extraction to enhance transparency and reproducibility. The objective of this review was to investigate blood-based transcriptomic immune responses in adult patients with CAP-associated sepsis, with particular emphasis on temporal immune trajectories and gene expression signatures associated with clinical outcomes. The clinical question was structured according to the Population-Exposure-Comparator-Outcome (PECO/PICO) framework:
Population: Adult patients with CAP-associated sepsis
Exposure/Intervention : Blood transcriptomic profiling or gene expression analysis
Comparator: Survivors versus non-survivors or patients with different disease severity
Outcomes: Temporal immune trajectories, gene expression signatures, and candidate biomarkers associated with clinical outcomes
Literature search strategy
The search strategy was developed according to PRISMA guidelines. Literature search was performed in medRxiv, PubMed, and the Cochrane Central Register of Controlled Trials (CENTRAL) from database inception to March 2026. The search strategy combined Medical Subject Headings (MeSH) and free-text terms using Boolean operators. The main search string included terms related to community-acquired pneumonia, sepsis, and transcriptomics (such as, “community-acquired pneumonia” OR “CAP” AND “sepsis” AND “transcriptome” OR “transcriptomic” OR “gene expression”). The search results were restricted to human studies published in English. Review articles, editorials, comments, and study protocols were excluded. The literature search and initial screening were independently performed by ST and NA, while SW verified the search results and resolved discrepancies. Data extraction was conducted by ST, with independent validation by NA.
Study selection
The selection process was performed in multiple sequential stages, including title and abstract screening, as well as full-text assessment. During the initial screening stage, studies were excluded if they were review articles, editorials, comments, or study protocols. Studies conducted in animal models or in vitro systems were also excluded. In addition, articles focusing on pneumonia without sepsis, studies addressing acute respiratory distress syndrome without sepsis, and studies reporting purely clinical or methodological outcomes without transcriptomic or gene expression data were excluded.
Studies were retained for full-text review if they involved patients with sepsis or community-acquired pneumonia (CAP) associated with systemic infection, included critically ill or intensive care unit (ICU) populations, and reported blood-based transcriptomic or gene expression analyses relevant to immune response dynamics. Full-text articles were assessed for eligibility based on predefined inclusion and exclusion criteria. Studies were included if they reported transcriptomic profiling with defined clinical outcomes and, when available, longitudinal or serial blood gene expression sampling.
Risk of bias assessment
Risk of bias was assessed using an adapted version of the Newcastle-Ottawa Scale (NOS) designed for transcriptomic observational studies. Four domains were evaluated: selection bias, confounding control, transcriptomic quality, and outcome assessment. Selection bias, confounding control, and outcome assessment were classified as low, moderate, or high risk of bias. In contrast, transcriptomic quality was evaluated as an indicator of methodological robustness, with higher scores reflecting better data quality, including sequencing methodology, normalization procedures, and reporting transparency. Overall study quality was determined by integrating risk-of-bias domains with transcriptomic methodological quality.
Data extraction
Data were retrieved from each included study as follows: Cohort characteristics, including sample size and infection type; transcriptomic platform and analytical methodologies; differentially expressed genes and enriched biological pathways; recognized immune endotypes or gene expression profiles; and correlations between transcriptomic results and clinical outcomes. When available, information on sampling timepoints and longitudinal transcriptomic measurements was also extracted to enable evaluation of temporal immune trajectories.
Data synthesis
A narrative synthesis analysis was therefore conducted due to the heterogeneity in study design, patient populations, and analytical methodologies. The findings were analyzed and integrated utilizing a pathway and immune-axis framework to identify the immunological processes across studies. Particular emphasis was placed on early inflammatory responses, interferon signaling pathways, adaptive immune suppression, immune checkpoint activation, and transcriptional signatures associated with survival or mortality.
Synthesis of temporal transcriptomic data
Temporal immune dynamics were synthesized by aligning findings across biological phases of sepsis rather than at exact chronological time points. Because definitions of baseline sampling varied across studies (such as hospital admission, ICU admission, or sepsis diagnosis onset), direct day-to-day comparisons were not possible. Therefore, temporal data were harmonized into approximate biological phases, including the early hyperinflammatory phase, the intermediate immune transition phase, and the later immune suppression or immunoparalysis phase.
Limitations of the evidence synthesis
The included studies varied in sample size, transcriptomic platforms, sampling timing, and definitions of clinical endpoints, which limited the ability to perform quantitative pooling. In addition, differences in baseline timepoint definitions (such as ICU admission versus hospital presentation) required temporal comparisons to be interpreted cautiously. Despite these limitations, the synthesis approach enabled identification of recurrent immune pathways, transcriptional signatures, and temporal immune patterns associated with CAP-associated sepsis outcomes.
Results
Study selection
The systematic literature search identified 607 records. After title and abstract screening, 588 articles were excluded for not meeting the predefined inclusion criteria (Figure 1). Nineteen studies were assessed through a full-text review, of which 6 met the eligibility criteria and were included in the final synthesis. The included studies were heterogeneous in design, cohort size, and transcriptomic platforms, necessitating a narrative synthesis of biological patterns rather than a quantitative meta-analysis.
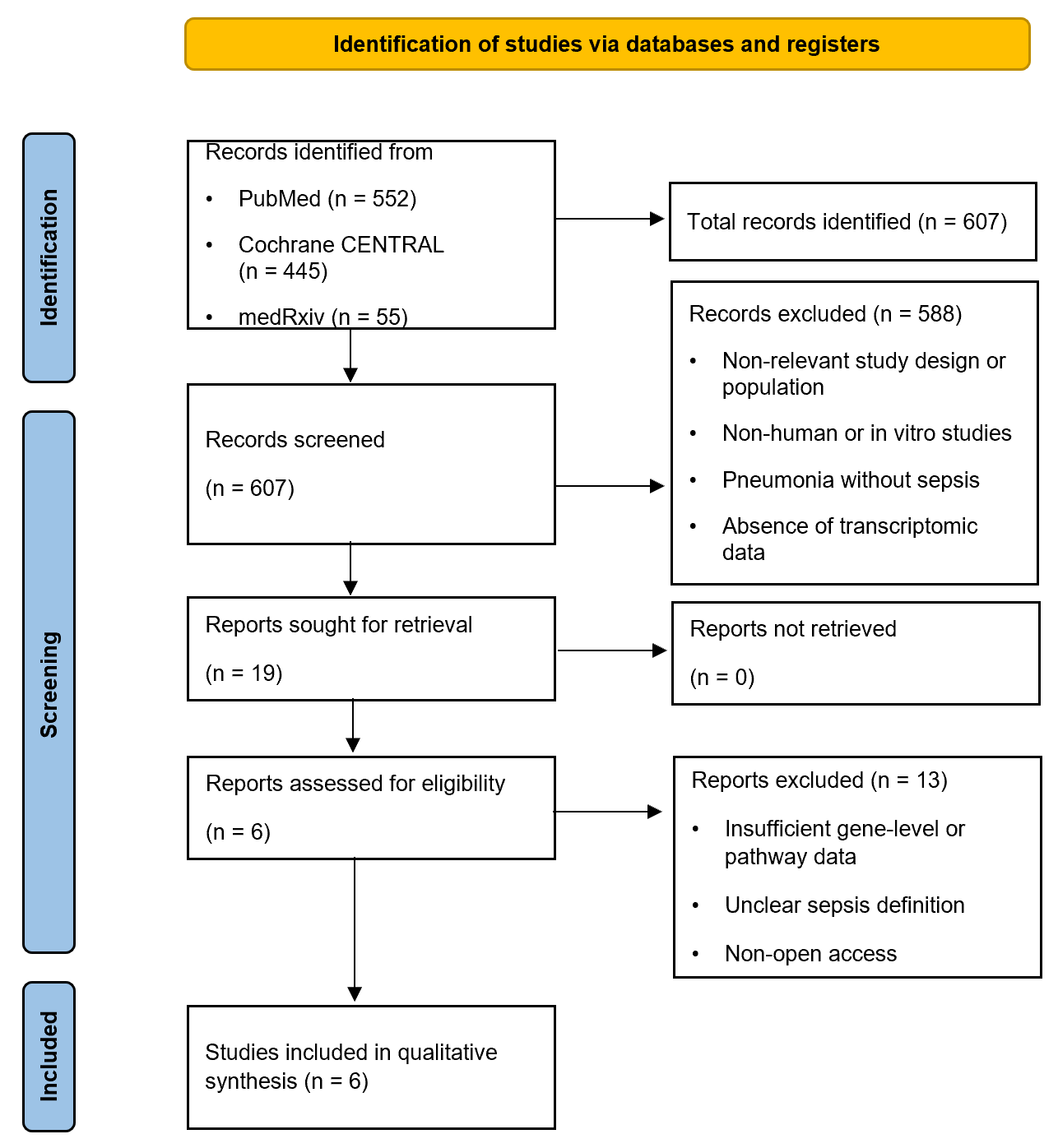
Figure 1 PRISMA 2020 flow diagram of the identification, selection and screening stages of the systematic reviews process.
Study characteristics and cohort structure
The demographic and clinical characteristics of sepsis cohorts from 3 studies are summarized in Table 1. These studies included critically ill adults with sepsis admitted to the intensive care unit (ICU). Disease severity was evaluated throughout the cohorts utilizing validated clinical criteria, such as Sequential Organ Failure Assessment (SOFA) and Acute Physiology and Chronic Health Evaluation II (APACHE II). Furthermore, one study reported details on survivors (n = 5) and non-survivors (n = 5), while another reported an intervention using anakinra (n = 263) and a placebo with standard care (n = 130). In addition, there was one study focused on sepsis with or without coronavirus diseases 2019 (COVID-19) (total n = 42). The primary clinical outcomes included survival and mortality.
Table 1 Data extraction from sepsis cohorts.
Study |
Population / Group |
N |
Age (years) |
Male (%) |
Severity score(s) |
Key outcomes |
Severino et al. [18] |
Sepsis survivors (CAP) |
5 |
47 (25 - 82)* |
NR |
APACHE II, SOFA |
Survival |
|
Sepsis non-survivors (CAP) |
5 |
83 (57 - 92)* |
NR |
APACHE II, SOFA |
Mortality |
Kyriazopoulou et al. [19] |
Standard of Care |
130 |
62 ± 12 |
60 |
SOFA |
Severe respiratory failure, 28-day mortality |
|
Anakinra + Standard of Care |
263 |
62 ± 12 |
58.6 |
SOFA |
Reduced severe respiratory failure, reduced mortality rate |
An et al. [20] |
ICU non-COVID-19 sepsis |
22 |
56 ± 17.1 |
72.7 |
APACHE II, SOFA |
ICU survival |
|
ICU COVID-19 sepsis |
20 |
64 ± 11.8 |
80 |
APACHE II, SOFA |
ICU survival |
* Median (range), calculated from individual-level data. NR = not reported.
Sepsis cohorts from specific sources, along with their etiologies, are shown in Table 2. Two studies included in this section compared transcriptomic responses between sepsis caused by different infection sources, including CAP and fecal peritonitis (FP), and between sepsis caused by various pathogens, including bacteria and viruses. The number of sepsis populations focused on CAP and FP were 73 and 64, with replication cohorts of 53 and 53, respectively. Adding replication cohorts made the immune endotypes and outcome relationships that were found more reliable. The APACHE II and SOFA scores were reported as clinical indicators. The mortality rate for about 28 days of observation was recorded. In addition, etiology-based sepsis was reported for bacterial (n = 22) and viral (n = 35) pathogens with APACHE II and SOFA severity scores.
Table 2 Data extraction from source-specific and etiology-based sepsis cohorts.
Source-specific sepsis (CAP vs fecal peritonitis), Burnham et al. [14] |
||||||||
Cohort |
Infection source |
N |
Age (years) |
Male (%) |
APACHE II |
SOFA |
Mechanical ventilation (%) |
28-day mortality (%) |
Discovery |
CAP |
73 |
60.6 ± 15.6 |
50.6 |
18.0 ± 6.2 |
6.8 ± 3.5 |
69.9 |
17.8 |
|
FP |
64 |
65.1 ± 17.5 |
43.7 |
15.0 ± 5.9 |
5.4 ± 4.0 |
53.1 |
12.5 |
Validation |
CAP |
53 |
68.4 ± 13.7 |
69.8 |
19.7 ± 6.1 |
6.2 ± 3.8 |
35.8 |
13.2 |
|
FP |
53 |
68.0 ± 12.7 |
58.5 |
15.7 ± 5.9 |
6.3 ± 3.5 |
39.6 |
13.2 |
Etiology-based sepsis (bacterial vs viral), Muratsu et al. [22] |
||||||||
Study |
Group |
N |
Age (years) |
Male (%) |
APACHE II |
SOFA |
|
|
Muratsu et al. [22] |
Bacterial sepsis |
22 |
77.5 (65.3 - 82.0) |
72.7 |
Yes |
Yes |
|
|
|
Viral (COVID-19) sepsis |
35 |
72 (59 - 76) |
68.6 |
Yes |
Yes |
|
|
|
Healthy controls |
15 |
55 (40.5 - 59.0) |
50 |
NA |
NA |
|
|
NA = Not Available.
Table 3 shows the population of COVID-19 (n = 322), other respiratory infections (influenza, sepsis, and septic shock) (n = 96), co-infections (bacterial/viral/fungal) (n = 84), and healthy controls (n = 72). These datasets showed large-scale, and multicenter transcriptomic resources.
Table 3 Data extraction from large-scale and multicenter transcriptomic resources.
Study |
Population |
Participants (N) |
Key notes |
Chew et al. [21]
|
COVID-19 |
322 |
Multinational (Australia, Europe, SE Asia); longitudinal sampling in subset |
Other respiratory infections (influenza, sepsis, septic shock) |
96 |
Pre-pandemic cohorts |
|
Co-infections (bacterial/viral/fungal) |
84 |
Mixed pathogen profiles |
|
Healthy controls |
72 |
Pre-2019 baseline |
Risk of bias assessment
The methodological quality of the included studies was assessed using the Newcastle-Ottawa Scale. The risk-of-bias assessment indicated generally acceptable methodological quality among the included studies (Table 4). Most studies demonstrated low to moderate risk of selection bias, reflecting appropriate cohort selection and study design. Confounding control varied across studies, with some studies showing moderate limitations due to incomplete adjustment for potential clinical variables. In contrast, transcriptomic quality was consistently rated as high across the included studies, indicating robust sequencing methodologies, appropriate data processing, and adequate reporting of transcriptomic analyses. Outcome assessment was generally categorized as low to moderate risk. Additionally, most studies were classified as having moderate overall risk of bias. Overall, most studies clearly described patient selection and transcriptomic analysis methods; however, several studies had limitations related to small sample sizes, heterogeneous sampling timepoints, and limited external validation.
Table 4 Risk of bias assessment of included studies using a modified Newcastle-Ottawa framework adapted for transcriptomic research.
No |
Study |
Selection Bias |
Confounding Control |
Transcriptomic Quality |
Outcome Assessment |
Overall Risk |
1 |
Severino et al. [18] |
|
|
|
|
|
2 |
Burnham et al. [14] |
|
|
|
|
|
3 |
An et al. [20] |
|
|
|
|
|
4 |
Kyriazopoulou et al. [19] |
|
|
|
|
|
5 |
Muratsu et al. [22] |
|
|
|
|
|
6 |
Chew et al. [21] |
|
|
|
|
|
|
|
|
NA |
|
Moderate |
|
|
|
|
Low |
|
High |
|
Note: Selection bias, confounding control, and outcome assessment were evaluated as risk-of-bias domains (low, moderate, or high). Transcriptomic quality represents methodological robustness of the sequencing and analysis procedures, where red indicates high transcriptomic quality, orange moderate quality, and yellow lower quality. Grey indicates not applicable or not reported.
Landscape of transcriptomic research designs
Table 5 provides an overview of the 6 included studies. It shows how the studies differed in terms of study design, patient demographics, primary comparison or exposure, and main transcriptomic insight. The research included gene expression by using microarray, bulk ribonucleic acid (RNA) sequencing, and combined messenger RNA (mRNA)- microRNA (miRNA) analysis. Although there was variation in methodology, all research concentrated on gene expression profiling in sepsis with CAP during the study and documented longitudinal or outcome-related immune responses, facilitating integrative comparisons across datasets.
Table 5 Overview of included transcriptomic studies.
Study |
Study design |
Study population (summary) |
Primary comparison/ exposure |
Main transcriptomic insight |
Severino et al. [18] |
Cohort study (microarray) |
Small CAP-associated sepsis cohort |
Survivors vs non-survivors over time |
Early metabolic and oxidative phosphorylation signatures distinguished outcomes; sustained immune defense programs associated with survival. |
Kyriazopoulou et al. [19] |
Post-hoc analysis of RCT (SAVE-MORE) |
Severe COVID-19 pneumonia |
Anakinra vs placebo |
IL-1 blockade stabilized adaptive immune endotypes and reduced progression to coagulopathic and respiratory failure states. |
An et al. [20] |
Prospective cohort (whole blood transcriptomics) |
ICU sepsis from distinct infection sources |
Fecal peritonitis vs CAP |
A core sepsis transcriptional program was source-independent; SRS endotypes predicted early mortality. |
Burnham et al. [14] |
Prospective observational cohort (RNA-Seq) |
Severe pulmonary sepsis in ICU |
COVID-19 vs non-COVID sepsis (longitudinal) |
Highlighted early antiviral transcriptional divergence converged by day 7, reflecting immune-trajectory normalization. |
Muratsu et al. [22] |
Prospective observational study (RNA-Seq + miRNA) |
Bacterial vs viral sepsis |
Etiology-based comparison |
Bacterial sepsis induced stronger Th1 pathway dysregulation than viral (COVID-19) sepsis. |
Chew et al. [21] |
Data descriptor (multicentre resource) |
Respiratory infections and sepsis |
Dataset generation and validation |
Provides a large, validated whole-blood transcriptomic resource enabling cross-disease immune trajectory analyses. |
Transcriptomic immune programs in sepsis
To integrate findings from studies across heterogeneous cohorts and platforms, transcriptomic changes were categorized into 7 roles, which are shown in Table 6. These roles represent recurrent biological programs reported across multiple studies and sepsis etiologies. Early hyperinflammation, characterized by upregulation of neutrophil activation and cytokine amplification, often dominated early disease stages and led to cytokine storms. Several key genes involved in this stage were tumor necrosis factor (TNF), interleukin-1 beta (IL1B), IL6, IL8, chemokine (C-X-C motif) ligand 2/3 (CXCL2/3), S100 calcium-binding protein A12 (S100A12), and Triggering Receptor Expressed on Myeloid Cells 1 (TREM1). However, during viral sepsis, type I interferon signaling is initially enriched but rapidly attenuates over time. This signaling increased the expression of key genes, including Interferon alpha-inducible protein 27 (IFI27), interferon-induced proteins with tetratricopeptide repeats 1-3 (IFIT1–3), 2′-5′-Oligoadenylate Synthetase 2 (OAS2), oligoadenylate synthetase-like (OASL), interferon induced with helicase C domain 1 (IFIH1), and sialic acid binding Ig-like lectin 1 (SIGLEC1).
Table 6 Integrated immune axes and transcriptomic trajectories associated with sepsis outcomes.
Role |
Key genes* |
Functional role |
Trajectory in survivors |
Trajectory in non-survivors |
Supporting study |
Innate hyperinflammatory response |
TNF, IL1B, IL6, IL8, CXCL2/3, S100A12, TREM1 |
Neutrophil activation, cytokine amplification |
Early induction followed by resolution (D7) |
Early induction with sustained activation in a subset |
[14,18-20] |
Type I interferon signaling |
IFI27, IFIT1-3, OAS2, OASL, IFIH1, SIGLEC1 |
Viral sensing and IFN-mediated defense |
Transient induction at D0, resolution by D7 |
Similar transient induction |
[14,20-22] |
Antigen presentation & myeloid–T cell crosstalk |
HLA-DRA/B1/B3, HLA-DQA1, CD74, LAMP3, CTSS |
Antigen presentation and adaptive priming |
Progressive recovery (D7) |
Persistently suppressed |
[14,18-22] |
Th1 and cytotoxic T-cell programs |
TBX21, IFNG, STAT4, IL12RB1/2, CXCR3 |
Pathogen clearance and cellular immunity |
Recovery of Th1-associated transcription |
Sustained suppression |
[18,20,22] |
Immune checkpoint and T-cell exhaustion |
PDCD1, CD274, LAG3, CTLA4, TIGIT |
Negative regulation of T-cell activation |
Transient upregulation |
Sustained upregulation |
[19,20,22] |
Immunothrombosis and endothelial dysfunction |
F5, CR1, ICAM1, CLEC5A, STAB1, complement factors |
Vascular injury and organ failure |
Partial normalization |
Persistent dysregulation |
[14,18,21] |
Post-transcriptional immune regulation (miRNA) |
miR-21, miR-146a |
Broad immune silencing via mRNA repression |
Weak or transient |
Strong and sustained |
[22] |
*Key genes were selected based on the commonly reported genes that increased in CAP-associated sepsis.
Both pathogens showed similar responses afterwards, which altered antigen presentation and led to Th1-T-cell immunity. Those responses showed an immune recovery process with sustained suppression strongly related to non-survival. The key genes related to antigen presentation and adaptive priming included human leukocyte antigen (HLA)-DRA/B1/B3, HLA-DQA1, cluster of differentiation 74 (CD74), lysosome-associated membrane protein 3 (LAMP3), and Cathepsin S (CTSS). Furthermore, the key genes associated with pathogen clearance and cellular immunity included T-box transcription factor 21 (TBX21), interferon Gamma (IFNG), signal transducer and activator of transcription 4 (STAT4), Interleukin-12 Receptor Beta Subunits 1 and 2 (IL12RB1/2), and CXCR3.
Immune checkpoints, driven by immune exhaustion, often co-occur with defects in antigen presentation and Th1 suppression, particularly in immunoparalysis. This condition is characterized by negative regulation of key genes involved in T-cell activation, including programmed cell death 1 (PDCD1), CD274, lymphocyte-activation gene 3 (LAG3), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), and T-cell immunoreceptor with Ig and ITIM domains (TIGIT). The upregulation of those genes suppressed effector T cells and impaired the immune response to eliminating the pathogens.
In addition, the persistent pathway linked to organ failure and mortality was dysregulated, as indicated by key genes such as coagulation Factor V (F5), complement receptor 1 (CR1), intercellular adhesion molecule 1 (ICAM1), C-type lectin domain family 5, member A (CLEC5A), and stabilin 1 (STAB1), which serve as markers an immunothrombosis and endothelial dysfunction. Finally, the high level of post-transcriptional immune regulation, such as miRNA, including miR-21 and miR-146a, weakened the recovery processes and reinforced long-term immune suppression in non-resolving sepsis. Collectively, these pathways provided a unified framework to interpret temporal immune trajectories and outcome-associated transcriptional programs across bacterial and viral sepsis.
Temporal immune dynamics in sepsis
Based on the immune axes summarized in Table 6, we next examined how these pathways evolve and diverge across the course of sepsis. Although sampling time points differed among studies, consistent transcriptional patterns allowed reconstruction of a shared immune trajectory from early hyperinflammation to adaptive immune dysregulation.
Early hyperinflammation phase
All studies included in the earliest transcriptomic data, collected on day 0 when the patient first arrived at the hospital or was admitted to the ICU, showed an innate hyperinflammatory response. The data showed the upregulation of TNF, IL1B, IL6, IL8, CXCL2, CXCL3, S100A12, and TREM1. These genes work together to bring in and activate neutrophils and monocytes, which shows that the innate immune response is strong at the start of the disease [18-20]. A similar inflammatory pattern was observed in both COVID-19 and non-COVID-19 sepsis groups [14], indicating that early hyperinflammation is a host response to systemic infection that is largely nonspecific to any single pathogen. Notably, this hyperinflammatory response was frequently more significant in survivors compared to non-survivors [18]. These results suggest that the highest early cytokine activation, which is often associated with early sepsis mortality [19,20], does not inherently predict adverse outcomes [18,20]. Instead, early inflammation seems to be temporary, and in people who survive, it goes away as part of a strong immune response that helps them get better [18,20]. Type I interferon (IFN-I) signaling, on the other hand, gave clear information about specific pathogens at early time points, unlike the nonspecific inflammatory response.
IFN-I expression was significantly elevated in viral sepsis relative to bacterial sepsis. The induction of IFN-stimulated genes (ISGs) such as IFI27, IFIT1-3, OAS2/OASL, IFIH1, and SIGLEC1 constituted a unique molecular signature of viral sepsis, particularly COVID-19, upon ICU admission [14,20,21]. However, this antiviral program did not express for a long time. By Day 7, the concentrations of IFN and ISG had significantly decreased, indicating a transition from an initial antiviral state to a more generalized immune-dysregulated phenotype. Consequently, although pathogen types influence initial transcriptional signatures, these differences rapidly diminish, leading in both viral and bacterial sepsis following the same immunological trajectory. These data highlight the importance of timing in sepsis diagnosis, as IFN-driven biomarkers such as IFI27 are highly informative during the acute phase but lose predictive value as the host response advances.
Adaptive suppression and Th1 collapse
Following the early inflammatory phase, a key point of clinical outcome was the inability to restore antigen presentation and adaptive immune signaling. The survivors demonstrated significant re-expression of major histocompatibility complex class II (MHC-II) pathway genes, including HLA-DRA, HLA-DRB1/3, HLA-DQA1, CD74, CTSS, and LAMP3 [18-20]. In contrast, continuous suppression of MHC-II genes in non-survivors reduced T-cell receptor (TCR) signaling and correlated with elevated expression of inhibitory receptors including PD-1, suggesting an extensive failure of adaptive immunity [20]. The restoration of MHC-II expression indicates a pivotal transition towards immune resolution. Importantly, the failure of antigen-presenting cell (APC) function was consistent across bacterial, viral, COVID-19, CAP, and mixed-sepsis groups, highlighting its pathogen-independent significance in lethal immune pathways.
The suppression of APC activity alters Th1 immunity, as indicated by downregulation of TBX21 (T-bet), IFNG, STAT4, IL12RB1, IL12RB2, and CXCR3 [22]. Although viral sepsis exhibited similar patterns, Th1 suppression was generally less pronounced, suggesting a more severe adaptive immune collapse in bacterial sepsis. Collectively, these findings describe an immune trajectory in which initial innate activation is followed by progressive adaptive immune decline, particularly in patients who fail to recover antigen presentation and Th1-mediated cellular immunity.
Immune checkpoint activation and T-cell exhaustion
Adaptive immune suppression was further reinforced by upregulation of inhibitory immune checkpoint pathways. Increased expression of PDCD1 (PD-1), CD274 (PD-L1), LAG3, CTLA4, and TIGIT indicated widespread T-cell exhaustion and impaired lymphocyte activation [22]. Similarly, the SRS1 endotype had more checkpoint and T-cell inhibitory modules, which showed that PD-1/PD-L1 signaling is a key transcriptional feature of immunoparalysis [19,20]. In a mechanistic way, checkpoint activation inhibits T-cell receptor signaling, induces lymphocyte anergy, and amplifies suppression of antigen presentation pathways. This creates a self-reinforcing cycle of adaptive immune dysfunction, linking early inflammation to later immune paralysis.
Vascular dysfunction
Beyond immune cell dysfunction, transcriptomic analysis consistently identified vascular and coagulation pathways as key contributors to sepsis pathophysiology. Coagulation-related gene signatures, including F5, CR1, ICAM1, CLEC5A, and STAB1, consistently correlated with organ dysfunction and poor outcomes in RNA-seq cohorts [21]. Both COVID-19 and non-COVID sepsis cohorts showed persistent coagulation, platelet degranulation, and complement activation pathways through Day 7, even after inflammatory and interferon responses had declined [14]. Survivors demonstrated partial restoration of vascular pathways, whereas non-survivors exhibited sustained dysregulation, underscoring the importance of vascular repair and hemostatic balance in recovery.
An additional regulatory layer involved post-transcriptional immune modulation mediated by microRNAs. Several studies reported extensive upregulation of miRNAs, including miR-21 and miR-146a, which collectively targeted thousands of mRNAs involved in antigen presentation, Th1 differentiation, and inflammatory signaling [22]. This miRNA-mediated repression contributed to prolonged immune suppression and persistent immunoparalysis, particularly in non-survivors and SRS1 endotypes. Based on this concept, we propose a putative model that integrates innate activation, antigen presentation, and adaptive immune responses in sepsis (Figure 2).
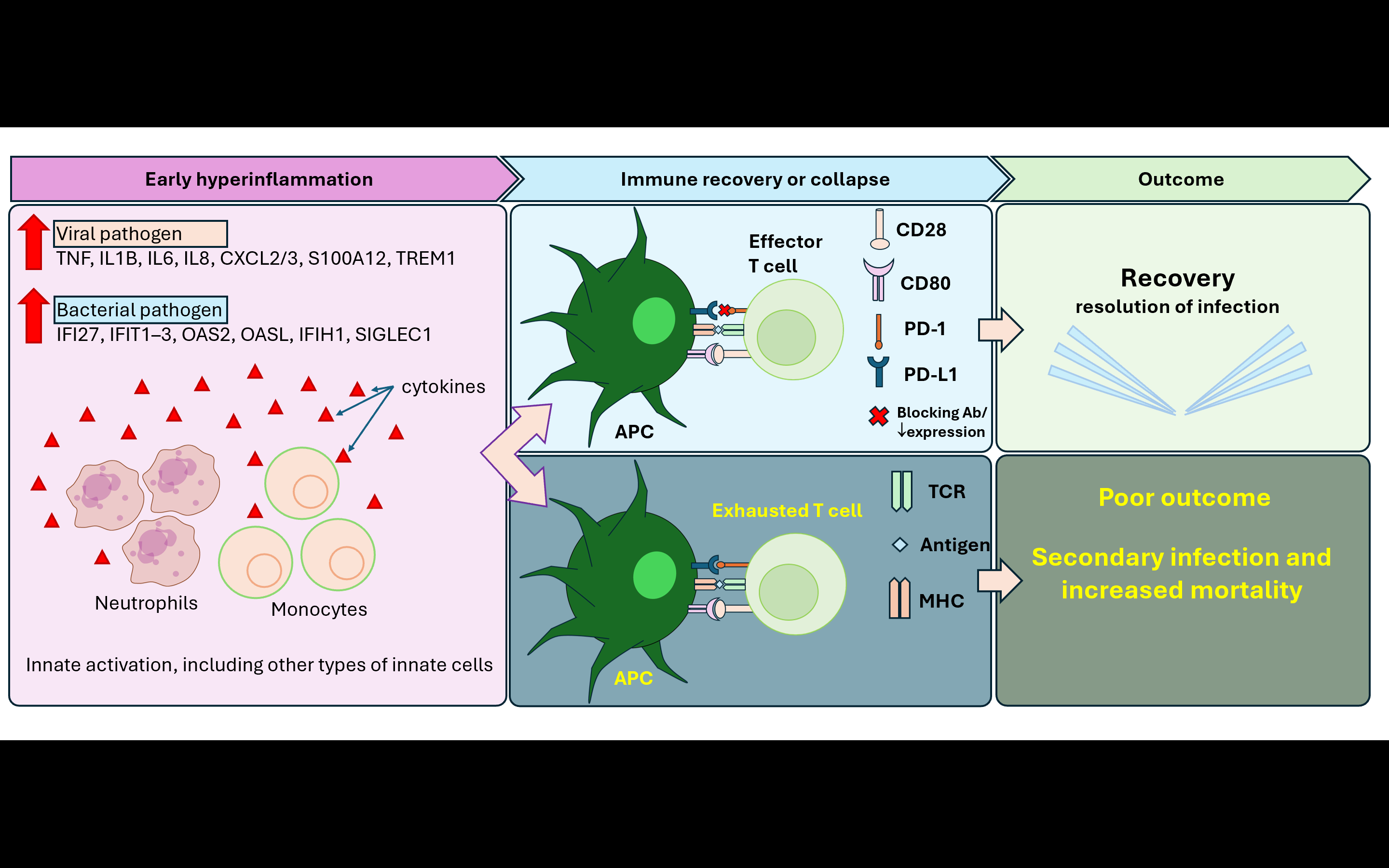
Figure 2 Proposed model of temporal immune trajectory in CAP-associated sepsis. This figure shows an integrative synthesis of transcriptomic findings across included studies. Early CAP-associated sepsis is marked by hyperinflammation, with pro-inflammatory cytokines gradually increasing, indicating activation of innate immune cells, including neutrophils and monocytes. There are 2 outcomes after this condition: either restoration or immune collapse. The recovery of antigen-presenting cell (APC) function and preserved co-stimulatory signaling (CD80-CD28) support effector T cell activation. In contrast, sustained APC dysfunction impaired antigen presentation and led to the dominance of inhibitory immune checkpoint signaling (PD-1/PD-L1), which is associated with T-cell exhaustion, resulting in adaptive immune collapse and poor outcomes, including increased risk of secondary infection and mortality.
We also propose a putative temporal model summarizing the divergence of immune responses during sepsis based on the integration of longitudinal transcriptomic patterns across the included studies (Figure 3). This model illustrates how early hyperinflammation may either resolve toward immune recovery or transition into prolonged immunosuppression, which is associated with secondary infections and increased late mortality.
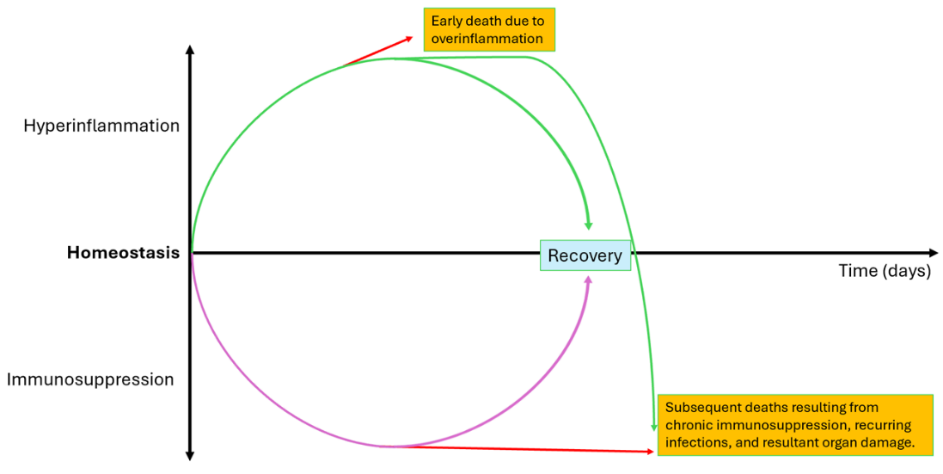
Figure 3 Proposed model of immune response and clinical outcomes in patients with CAP-associated sepsis. This model illustrates hypothesized trajectories of immune responses based on included studies in patients with CAP-associated sepsis. The studies included showed that patients with CAP-associated sepsis are disturbed in immune homeostasis, as indicated by an early hyperinflammatory phase that may progress into the recovery phase. However, in some patients, uncontrolled hyperinflammation leads to early mortality. In patients who survive the initial phase of hyperinflammation, the immune response may gradually return to normal, leading to clinical recovery. However, in non-survivors who passed the early hyperinflammation phase, they may experience prolonged immune dysregulation, progressing by sustaining immunosuppression, leading to progressive organ damage, resulting in the risk of mortality.
Candidate biomarkers
Across the included studies, several genes consistently emerged as key transcriptomic markers of the sepsis immune response. We highlighted representative genes that reflect temporal phases of specific immune responses observed across studies, as shown in Table 7. IL1B and TREM1, both genes related to innate immune activation, were frequently upregulated in sepsis patients and were associated with inflammation driven by myeloid cells.
Table 7 Candidate biomarkers representing conserved immune response in sepsis.
Biomarker |
Immune Axis |
Association |
Functional Interpretation |
Reason |
IL1B |
Early hyperinflammation |
Acute inflammatory response |
Pro-inflammatory cytokine driving innate activation |
Consistently upregulated across sepsis cohorts |
TREM1 |
Myeloid amplification |
Innate immune activation |
Neutrophil and myeloid inflammatory signaling |
Robust marker of myeloid activation |
IFI27 |
Type I interferon |
Viral-associated response |
IFN-stimulated gene |
The expression is increased in the virus than in the bacteria. |
HLA-DRA |
Antigen presentation |
Immune competence vs immunoparalysis |
MHC-II expression reflecting APC functionality |
Gold-standard marker of immunoparalysis |
TBX21 |
Th1 immunity |
Adaptive immune integrity |
Master regulator of Th1 differentiation |
Central indicator of Th1-cell functional collapse |
PDCD1 |
Immune exhaustion |
Persistent immune dysfunction |
Immune checkpoint mediating T-cell inhibition |
Associated to non-survival |
In both pathogens, IFI27, a type I interferon, was strongly elevated by viral compared to bacterial pathogens, particularly in COVID-19, supporting its role in clearing viral pathogens. Furthermore, the innate immune response and interferon cytokines altered antigen presentation and effector T cell function. HLA-DRA and TBX21 were suppressed, suggesting immunoparalysis or non-survival phenotypes that reflect impaired antigen presentation and adaptive immune competence, especially in Th1 differentiation.
Another key gene that has important implications in immune checkpoint genes was PDCD1. Increased PDCD1 expression indicates immune dysfunction, suggesting a mechanistic link between antigen failure and T-cell exhaustion. Overall, those genes represent distinct yet interrelated immunological axes that characterize the pathophysiology of sepsis and provide a rational basis for focused validation as transcriptomic biomarkers.
Discussion
Our synthesis showed that early hyperinflammation in CAP-associated sepsis initiates divergent adaptive immune trajectories, resulting in either effective effector T-cell activation or progressive T-cell exhaustion. These findings are relevant to CAP-associated sepsis, which is characterized by early clinical decline coming after late-stage immune dysfunction. Early hyperinflammation was characterized by rapid activation of innate immune sensing pathways and circulating leukocyte reprogramming, consistent with classical descriptions of sepsis-induced dysregulated host responses. The elevated cytokine levels indicated rapid activation of neutrophils and inflammatory monocytes, leading to hyperinflammation. This early phase reflects pattern-recognition receptor–mediated sensing of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by innate immune cells, triggering downstream inflammatory cascades [23,24].
Interestingly, the cytokine storm in early hyperinflammation occurred independently of the pathogen. Such pathogen-independent inflammatory activation has been widely described as a hallmark of early sepsis, driven by convergent innate immune pathways [25]. However, this type of cytokine storm could not predict the outcome of sepsis. Although early death is often associated with excessive inflammation, this review found that survivors usually exhibit equivalent or even stronger early cytokine production. This suggests that early hyperinflammation does not always indicate adverse outcomes. Instead, early inflammation appears to represent an intact and responsive innate immune system, while subsequent immune regulation determines prognosis. Instead, the immunological balance and recovery occur after this initial innate reaction, which is the crucial factor.
Our findings suggest that the survivor restored antigen-presenting cells (APCs) function. APCs, including monocytes, dendritic cells, and B cells, are essential for connecting innate sensing to adaptive effector T cells, supporting sepsis recovery [26]. During sepsis, the APC become depleted and weakened, altering the T cell priming and immune balance. Therefore, DC recovery in survivors is more likely to reflect maintained or regained APC functionality, whereas in non-survivors reduced antigen presentation leads to immunoparalysis in sepsis.
The paralysis of APC inhibited adaptive T-cell activation by restricting TCR signaling. Impaired antigen presentation and reduced HLA-DR expression on monocytes and DCs are well-established hallmarks of septic immunosuppression, directly limiting TCR engagement and adaptive immunity [27]. Sepsis altered lymphocyte apoptosis and exhaustion, mainly affecting CD4+ and CD8+ T cells [28]. Recent studies showed that regulatory T cells (Tregs) proliferate proportionately during sepsis to balance the effector and regulatory lymphocyte populations. Tregs regulate immune response by producing IL-10 and transforming growth factor beta (TGF-β), which directly inhibit the effector T cells [29]. In the event of significant effector T-cell loss, Treg expansion is likely to strengthen immunosuppression rather than resolve it [29].
The polarization of macrophages is crucial for reprogramming the sepsis niche around injured tissue by switching macrophage phenotypes toward type 2 macrophages (M2). M2 reduced the pro-inflammatory cytokine secretion, affected phagocytosis, and altered antigen presentation [30]. This condition enhanced tissue remodeling, whereas in the non-survivor, HLA-DR was downregulated on monocytes. Furthermore, the dynamic of immune response from the early hyperinflammation to recovery influenced both innate and adaptive immunity. Natural killer (NK) cells are innate immune cell that produce IFN-γ and modulate the Th1 effector response [31,32]. Thus, the decreased Th1 responses in non-survivors suggest that it may be indirectly caused by NK dysfunction. IFN-γ is an essential modulator of early macrophage activation and Th1 polarization.
Another convergent process underlying adaptive immune failure is the activation of immunological checkpoints. Receptors such as PD-L1 on lymphocytes and its ligand, PD-1, contribute to suppressing effector T cells, making them exhausted [29]. Our review identified overexpression of PDCD1 (PD-1), CD274 (PD-L1), CTLA4, LAG3, and TIGIT, findings in line with functional studies on the SRS1 endotype and immunoparalysis. This inhibition further suppresses TCR signaling and strengthens antigen-presentation failure, leading to immune dysfunction.
Alongside immune signaling, vascular and homeostatic dysregulation further distinguishes the fatal processes. Vascular endothelial cells and platelets collaborate to modulate sepsis pathology by releasing DAMPs and amplifying inflammatory injury, relating immune activation to organ dysfunction. Non-survivors continue to activate the coagulation, platelet degranulation, and complement pathway [24]. Organ failure, microvascular thrombosis, and endothelial damage have been linked to genes such as ICAM1, F5, CR1, and CLEC5A.
In addition, immune suppression is achieved through post-transcriptional regulation. microRNAs such as miR-21 and miR-146a suppress antigen presentation, Th1 differentiation, and inflammatory signaling, leading to adaptive immune dysfunction even after cytokine-storm levels have decreased. Overall, these findings showed that gradual immunological paralysis or immune recovery occurred after early innate hyperactivation. This dynamic immune trajectory involves a shift from early inflammation to late immunosuppression. Our findings and the existing literature highlight the importance of an immunomodulatory strategy in suppressing inflammation, tailored to immunological timing and endotype. Despite these insights, several limitations should be considered when interpreting the findings of this review. First, only 6 studies met the inclusion criteria, reflecting the limited availability of longitudinal transcriptomic datasets specifically focusing on CAP-associated sepsis. Second, heterogeneity in study design, patient populations, and sampling timepoints across the included studies limited the possibility of quantitative synthesis and direct comparison of temporal immune trajectories. In addition, although the overall methodological quality of the included studies was considered moderate, several studies had relatively small sample sizes and limited external validation of transcriptomic signatures. These methodological differences may influence the generalizability of the identified immune trajectories and candidate biomarkers.
Although whole-blood transcriptomic profiling provides valuable mechanistic insights into immune dysregulation during sepsis, its real-time implementation in intensive care settings remains challenging. Current RNA sequencing workflows require specialized infrastructure and often involve turnaround times exceeding 24 - 48 h, limiting their utility for immediate clinical decision-making. Furthermore, the cost and technical expertise required for high-throughput sequencing may limit its routine use in many clinical settings.
Emerging technologies, including targeted gene-expression panels and rapid multiplex platforms such as NanoString and PCR-based assays, may provide more practical alternatives. These approaches allow focused quantification of selected immune-related transcripts and may enable faster turnaround times compatible with ICU workflows. However, prospective validation studies are required to determine whether simplified transcriptomic signatures can reliably guide clinical management in CAP-associated sepsis.
Conclusions
This review synthesizes current evidence on temporal immune trajectories in CAP-associated sepsis and identifies several candidate transcriptomic biomarkers associated with disease progression and clinical outcomes. These findings suggest that dynamic immune signatures may provide insights into patient stratification and potential targets for immunomodulatory strategies. However, further prospective studies with standardized longitudinal sampling are required to validate these biomarkers and clarify their clinical utility.
Acknowledgements
This review was funded by Universitas Padjadjaran through the Indonesian Endowment Fund for Education (LPDP) on behalf of the Indonesian Ministry of Higher Education, Science and Technology, and was managed under the EQUITY Programme (Contract Nos. 4303/83/DT.03.08/2025 and 3927/UN6.RKT/HK.07.00/2025) for NA.
Declaration of generative AI in scientific writing
This manuscript preparation used AI-assisted language to improve grammar, clarity, and readability under full human supervision. The scientific content and interpretations are the responsibility of the authors.
CRediT author statement
Suparto: Conceptualization; Methodology; Visualization; Formal analysis; writing-original draft. Suwarman: Writing - Review & Editing; Investigation. Wani Devita Gunardi: Formal analysis; Writing - Review & Editing; Supervision. Henny Tannady Tan: Writing - Review & Editing; Project administration. Nathalie Widjaja: Writing - Reviewing and Editing; Methodology. Nur Atik: Conceptualization; Methodology; Writing - Review; Editing and Supervision.
References
[1] JP Metlay, GW Waterer, AC Long, A Anzueto, J Brozek, K Crothers, LA Cooley, NC Dean, MJ Fine, SA Flanders, MR Griffin, ML Metersky, DM Musher, MI Restrepo and CG Whitney. Diagnosis and treatment of adults with community-acquired pneumonia. An official clinical practice guideline of the American Thoracic Society and Infectious Diseases Society of America. American Journal of Respiratory and Critical Care Medicine 2019; 200(7), e45-e67.
[2] MN Lutfiyya, E Henley, LF Chang and SW Reyburn. Diagnosis and treatment of community-acquired pneumonia. American Family Physician 2006; 73(3), 442-450.
[3] J Womack and J Kropa. Community-acquired pneumonia in adults: Rapid evidence review. American Family Physician 2022; 105(6), 625-630.
[4] MN Lutfiyya, E Henley, LF Chang and SW Reyburn. Diagnosis and treatment of community-acquired pneumonia. American Family Physician 2006; 73(3), 442-450.
[5] SM Han, PS Hon, HY Na, TTH Yong, PA Tambyah and YT Wen. Viral non-SARS-CoV-2 etiology of community-acquired pneumonia (CAP) in Southeast Asia: a review and pooled analysis. IJID Regions 2025; 15, 100672.
[6] Z Hu, J Lin, J Chen, T Cai, L Xia, Y Liu, X Song and Z He. Overview of viral pneumonia associated with influenza virus, respiratory syncytial virus, and coronavirus, and therapeutics based on natural products of medicinal plants. Frontiers in Pharmacology 2021; 12, 630834.
[7] W Xu, Y Zhang, Y Gao and X Li. Impact of the corona virus disease 2019 pandemic on the prevalence of common respiratory pathogens in hospitalized young patients with non-severe community-acquired pneumonia. Journal of Thoracic Disease 2025; 17(10), 107216.
[8] V Biradar and JL Moran. SIRS, sepsis and multiorgan failure. In: R Fitridge and M Thompson (Eds.). Mechanisms of vascular disease: A reference book for vascular specialists. University of Adelaide Press, South Australia, Australia, 2011.
[9] L La Via, G Sangiorgio, S Stefani, A Marino, G Nunnari, S Cocuzza, I La Mantia, B Cacopardo, S Stracquadanio, S Spampinato, S Lavalle and A Maniaci. The global burden of sepsis and septic shock. Epidemiologia 2024; 5(3), 456-478.
[10] Y Li, S Ren and S Zhou. Advances in sepsis research: Insights into signaling pathways, organ failure, and emerging intervention strategies. Experimental and Molecular Pathology 2025; 142, 104963.
[11] MA Ovali and S Percin. Sepsis-associated immunosuppression: Mechanistic insights, biomarkers, and therapeutic perspectives. Molecular Biology Reports 2025; 53(1), 148.
[12] L Stiel, A Gaudet, S Thietart, H Vallet, P Bastard, G Voiriot, M Oualha, B Sarton, H Kallel, N Brechot, L Kreitmann, S Benghanem, J Joffre and Y Jouan. Innate immune response in acute critical illness: A narrative review. Annals of Intensive Care 2024; 14(1), 137.
[13] X Tao, J Wang, B Liu, P Cheng, D Mu, H Du and B Niu. Plasticity and crosstalk of mesenchymal stem cells and macrophages in immunomodulation in sepsis. Frontiers in Immunology 2024; 15, 1338744.
[14] KL Burnham, EE Davenport, J Radhakrishnan, P Humburg, AC Gordon, P Hutton, E Svoren-Jabalera, C Garrard, AVS Hill, CJ Hinds and JC Knight. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. American Journal of Respiratory and Critical Care Medicine 2017; 196(3), 328-339.
[15] JG Chenoweth, J Brandsma, DA Striegel, P Genzor, E Chiyka, PW Blair, S Krishnan, E Dogbe, I Boakye, GB Fogel, EL Tsalik, CW Woods, A Owusu-Ofori, C Oppong, G Oduro, T Vantha, AG Letizia, CG Beckett, KL Schully and DV Clark. Sepsis endotypes identified by host gene expression across global cohorts. Communications Medicine 2024; 4(1), 120.
[16] RE Hancock, A An, CC dos Santos and AH Lee. Deciphering sepsis: Transforming diagnosis and treatment through systems immunology. Frontiers in Science 2025; 2, 1469417.
[17] YY Zhang and BT Ning. Signaling pathways and intervention therapies in sepsis. Signal Transduction and Targeted Therapy 2021; 6, 407.
[18] P Severino, E Silva, GL Baggio-Zappia, MKC Brunialti, LA Nucci, O Rigato, IDCG da Silva, FR Machado and R Salomao. Patterns of gene expression in peripheral blood mononuclear cells and outcomes from patients with sepsis secondary to community acquired pneumonia. PLoS One 2014; 9(3), e91886.
[19] E Kyriazopoulou, Y Hasin-Brumshtein, U Midic, G Poulakou, H Milionis, S Metallidis, M Astriti, A Fragkou, A Rapti, E Taddei, I Kalomenidis, G Chrysos, A Angheben, I Kainis, Z Alexiou, F Castelli, FS Serino, P Bakakos, E Nicastri, …, EJ Giamarellos-Bourboulis. Transitions of blood immune endotypes and improved outcome by anakinra in COVID-19 pneumonia: An analysis of the SAVE-MORE randomized controlled trial. Critical Care 2024; 28, 73.
[20] AY An, A Baghela, P Zhang, R Falsafi, AH Lee, U Trahtemberg, AJ Baker, CC dos Santos and REW Hancock. Severe COVID-19 and non-COVID-19 severe sepsis converge transcriptionally after a week in the intensive care unit, indicating common disease mechanisms. Frontiers in Immunology 2023; 14, 1167917.
[21] T Chew, TM Pelaia, AL Phu, S Teoh, Y Wang, N Deshpande, K Kim, V Herwanto, Gunawan, T Karvunidis, Y Zerbib, KR Short, S Macdonald, I Thevarajan, D Rinchai, WS Kuan, B Knippenberg, J Iredell, PN Britton and M Shojaei. Molecular landscape of respiratory infection: A large-scale, multi-centre blood transcriptome dataset. Scientific Data 2025; 12(1), 1175.
[22] A Muratsu, S Oda, S Onishi, J Yoshimura, H Matsumoto, Y Togami, Y Mitsuyama, H Ito, D Okuzaki, H Ogura and J Oda. Bacterial sepsis causes more dramatic pathogenetic changes in the Th1 pathway than does viral (COVID-19) sepsis: A prospective observational study of whole blood transcriptomes. Virology Journal 2024; 21(1), 190.
[23] WJ Wiersinga, SJ Leopold, DR Cranendonk and T van der Poll. Host innate immune responses to sepsis. Virulence 2014; 5(1), 36-44.
[24] T van der Poll, FL van de Veerdonk, BP Scicluna and MG Netea. The immunopathology of sepsis and potential therapeutic targets. Nature Reviews Immunology 2017; 17(7), 407-420.
[25] KH Lee, A Gordon and B Foxman. The role of respiratory viruses in the etiology of bacterial pneumonia. Evolution, Medicine, and Public Health 2016; 2016(1), 95-109.
[26] MJ Delano and PA Ward. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunological Reviews 2016; 274(1), 330-353.
[27] C Nedeva. Inflammation and cell death of the innate and adaptive immune system during sepsis. Biomolecules 2021; 11(7), 1011.
[28] F Franco, A Jaccard, P Romero, YR Yu and PC Ho. Metabolic and epigenetic regulation of T-cell exhaustion. Nature Metabolism 2020; 2(10), 1001-1012.
[29] J Brady, S Horie and JG Laffey. Role of the adaptive immune response in sepsis. Intensive Care Medicine Experimental 2020; 8(1), 20.
[30] CM Padovani and K Yin. Immunosuppression in sepsis: Biomarkers and specialized pro-resolving mediators. Biomedicines 2024; 12(1), 175.
[31] F Wang, Y Cui, D He, G Gong and H Liang. Natural killer cells in sepsis: Friends or foes? Frontiers in Immunology 2023; 14, 1011111.
[32] L Chiche, JM Forel, G Thomas, C Farnarier, F Vely, M Blery, L Papazian and E Vivier. The role of natural killer cells in sepsis. BioMed Research International 2011; 2011(1), 986491.