Trends
Sci.
2026;
23(9):
13123
Harnessing Natural Aryl Aldehydes to Synthesize Bromochalcones as Anticancer Candidates Targeting Proliferative Pathways
Amri
Setyawati1,2,
![]() ,
Niko Prasetyo1,
,
Niko Prasetyo1,
![]() ,
Indriana Kartini1,
,
Indriana Kartini1,
![]() ,
,
Endang
Astuti1,
![]() and Tutik Dwi Wahyuningsih1,*,
and Tutik Dwi Wahyuningsih1,*,
![]()
1Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada,
Yogyakarta 55581, Indonesia
2Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Islam Indonesia,
Sleman 55584, Indonesia
(*Corresponding author’s e-mail: [email protected])
Received: 23 December 2025, Revised: 30 January 2026, Accepted: 6 February 2026, Published: 5 April 2026
Abstract
Current cancer therapies are limited by sustainability, drug resistance, and off-target effects. Bromochalcones are suitable candidates for targeted therapies, owing to their designable structure that can engage proliferative pathways. This study aimed to synthesize bromochalcones from natural aryl aldehydes and to evaluate their structural features and in vitro and in silico anticancer properties. Six bromochalcones were synthesized and identified by FTIR, GC-MS, and NMR. The in vitro MTT assay showed that all brominated chalcones were more cytotoxic toward HeLa cells than toward MCF-7, T47D, and Vero cells. Compound (E)-1-(4-bromophenyl)-3-(4-hydroxy-3-methoxyphenyl) prop-2-en-1-one (referred to 2D), derived from vanillin, achieved the best results (IC50 = 5 µg/mL; SI = 90.71), outperforming doxorubicin (IC50 = 15 µg/mL). Molecular docking studies on several proliferation proteins indicate that PCNA had the highest correlation between binding affinity and the in vitro IC50 trend. 2D-PCNA complex exhibited the strongest binding affinity by forming hydrogen bonds with Glu25, Met40, and His44. A 100 ns molecular dynamics simulation also shows a stable interaction with a strong binding affinity. Reactivity analysis from DFT calculations indicates that bromochalcones 2A and 2D were the 2 most potent compounds in driving interactions, including hydrogen bonding. Furthermore, ADMET predictions assessment reveals the safety and potential of these compounds as drug candidates. In conclusion, vanillin-derived bromochalcone, 2D, emerges as the lead compound, demonstrating the best in vitro anticancer activity, a favorable in silico profile, and the highest potential for development into various formulations.
Keywords: Brominated-chalcones, IC50, Docking, Molecular dynamics, PCNA, DFT, ADMET
Introduction
Cancer remains a significant global health, economic, and social burden, with breast and cervical cancers consistently showing higher incidence and mortality [1]. The existing treatments, such as chemotherapy, radiotherapy, and targeted therapy, have limitations related to cell selectivity, severe side effects, and drug resistance [2]. Even promising candidates fail due to low solubility, instability, or rapid metabolism. Natural products have served as a source of anticancer drugs, including paclitaxel and vinblastine [3,4]. Natural
medicines are claimed to be safer than synthetic drugs, but their availability is limited due to difficulties in isolation [4]. Doxorubicin, a synthetic drug for chemotherapy, can provide a larger supply. However, it is limited by high toxicity and unselectivity, which often lead to more severe symptoms [5]. These have driven interest in semi-synthetic drugs, which leverage the best of both worlds by modifying natural scaffolds to maintain safety and scalability while enhancing pharmaceutical properties [6].
As intermediates in flavonoid biosynthesis [7], natural chalcones such as xanthohumol and isoliquiritigenin exhibit anticancer activity but are limited by low natural abundance and structural inflexibility [8-10]. Synthetic chalcones have also been developed and shown to possess anticancer activity. Nevertheless, many synthetic chalcones suffer from suboptimal bioavailability and extensive metabolism [11], limited selectivity with toxicity toward non-tumoral cells [12], and susceptibility to resistance or off-target effects [13-15]. Additionally, increasing environmental regulations (e.g., EPA, REACH) and the imperative for green chemistry strongly underscore the need to identify sustainable precursors [15]. To address this challenge, we turned to natural aryl aldehydes—including vanillin, anisaldehyde, veratraldehyde, cuminaldehyde, and cinnamaldehyde —as sustainable precursors for bromochalcone synthesis. These precursors provide diverse aromatic substituents (hydroxy, methoxy, and alkyl) that are essential for pharmacophore design and enhance anticancer activity [14]. All 6 precursors are also considered safe (GRAS), and admissible for use in food items, and accepted by the Flavor and Extract Manufacturers’ Association (FEMA) and the FDA. The GRAS status evaluation of aromatic aldehydes is relevant not only to food safety but also to the rational selection of anticancer drug precursors. A clear toxicological profile accelerated drug development by lowering early systemic toxicity risk.
Chalcones, especially bromochalcones, are a promising semi-synthetic scaffold for targeted cancer therapy due to their synthetic accessibility and structural versatility. Among these scaffolds, bromochalcones serve as a leading pharmacophore for developing targeted therapies against several key proteins. The phenyl bromide substituent fits into the binding pocket of succinate dehydrogenase [16]. Also, it enhances anticancer activity by interacting with the arginine residue on several cancer receptors, such as HER-2 and EGFR [17]. Although the structure–activity relationships of chalcone derivatives have been widely reviewed [18], and cinnamaldehyde-derived chalcones with anticancer activity have been reported [16], systematic exploration of brominated chalcones derived from diverse natural aryl aldehydes remains limited. Although a comprehensive review has explored the structure-activity relationship of chalcone derivatives as anticancer agents, a recent study reported the synthesis of cinnamaldehyde-derived chalcones with anticancer potential. Previous studies have reported that chalcone derivatives modulate protein pathways involved in proliferation. Chalcone has been shown to inhibit IGF1R in MCF-7 cells [19], and another chalcone analog has been shown to reduce Ki67 expression in a breast cancer model [20]. However, research on bromochalcone derivatives derived from other natural aldehydes that specifically target PCNA remains limited. PCNA has been recognized as a promising therapeutic target in cancer due to its central role in DNA replication and repair [21]. However, small-molecule inhibitors based on chalcone-derived scaffolds, particularly brominated variants derived from renewable natural aldehydes, have been far less explored, highlighting an opportunity to develop more selective and sustainable PCNA-targeting agents.
Herein, integrating experimental and computational approaches, we report the synthesis, characterization, and antiproliferative evaluation of bromochalcones derived from natural aryl aldehydes, targeting breast and cervical cancer cells with enhanced selectivity and therapeutic potential. We also evaluated their potency, selectivity, and binding mechanisms using DFT, docking, and molecular dynamics (MD). Furthermore, ADMET predictions were gathered to assess pharmacokinetics and safety.
Materials and methods
The p.a. materials for synthesis and MTT test, including doxorubicin and cisplatin, were purchased from Sigma-Aldrich, China. Thin-layer chromatography (TLC) was performed using aluminum plates (Merck) coated with silica gel 60 F-254 (20×20 cm2). HeLa, MCF-7, T47D cancer cells, and normal Vero cells were obtained from the Parasitology Laboratory at the Faculty of Medicine, Public Health, and Nursing, Gadjah Mada University.
The general synthesis involved refluxing 5 mmol of an aromatic aldehyde (1a-f), 5 mmol of 4-bromacetophenone, and 20% NaOH for 1 - 2 h. Progress was monitored using TLC and UV visualization. The reaction was halted by pouring into ice-cold water and acidifying with 10% HCl until a precipitate formed. The product was then recrystallized from ethanol and dried in a desiccator. The uncorrected melting and boiling points were measured using an Electrothermal 9100. Several spectra were obtained by FTIR (Shimadzu Prestige-21) and NMR (¹H at 500 MHz JEOL JNM ECA 500, with TMS as the internal standard, and CDCl₃ as solvent). Purity was confirmed by GC-MS (GC Agilent 6890 and Shimadzu QP 2010S).
Synthesis of (E)-1-(4-bromophenyl)-3-phenylprop-2-en-1-one (2A)
Bromochalcone 2A was synthesized via a general procedure using benzaldehyde as a precursor. It produces white powder in 68% yield. The purity is 99%, as indicated by a mass-to-charge ratio (m/z) of 287 in MS (EI), with a melting point (m.p.) of 92 °C and a boiling point (b.p.) of 146 °C. IR (ATR): νmax 1,658, 1,589, 1,512, 1,416, 1,257, 1,033, 990, 808 and 550 cm–1. The 1H-NMR shows the proton at δ 7.88 (d, J = 10 Hz, 2H), δ 7.80 (d, J = 15 Hz, 1Hα), δ 7.78 (d, J = 10 Hz, 1H), δ 7.63 (d, J = 10 Hz, 2H), δ 7.56 (d, J = 10 Hz, 1H), δ 7.46 (d, J = 15 Hz, 1Hβ), δ 7.41 (d, J = 10 Hz, 2H) and δ 7.41 (m, J = 10 Hz, 1H).
Synthesis of(E)-1-(4-bromophenyl)-3-(4-methoxyphenyl) prop-2-en-1-one (2B)
Bromochalcone 2B was synthesized via a general procedure using p-anisaldehyde as a precursor. It produces pink powder in 89% yield. The purity is 99%, as indicated by a mass-to-charge ratio (m/z) of 318 in MS (EI), with a melting point (m.p.) of 128 °C and a boiling point (b.p.) of 270 °C. IR (ATR): νmax 3,248, 2,954, 1,681, 1,608, 1,581, 1,450, 1,334, 1,211, 1,072, 987, 825 and 763 cm–1.The 1H-NMR shows the proton at δ 7.86 (d, J = 10 Hz, 2H), δ 7.76 (d, J = 15 Hz, 1Hα), δ 7.61 (d, J = 10 Hz, 2H), δ 7.57 (d, J = 10 Hz, 2H), δ 7.33 (d, J = 15 Hz, 1Hβ), δ 6.91 (d, J = 10 Hz, 2H) and δ 3.93 (s, 3H).
Synthesis of (E)-1-(4-bromophenyl)-3-(3,4-dimethoxyphenyl) prop-2-en-1-one (2C)
Bromochalcone 2C was synthesized via a general procedure with veratraldehyde as a precursor. It produces greenish powder in 55% yield. The purity is 95%, as determined by MS (EI) at m/z 348, with m.p.: 76 °C and b.p.: 140 °C. IR (ATR): νmax 2,934, 2,834, 1,672, 1.583, 1,515, 1,458, 1,259, 1,071, 1,026, 979, 809 and 771 cm–1. The 1H-NMR shows the proton in δ 7.80 (d, J = 35.0 Hz, 1H), 7.60 (d, J = 16.0 Hz, 1Hα), 7.39 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 8.8 Hz, 1H), 7.22 (d, J = 14.0 Hz, 1Hβ), 7.14 (d, J = 8.3 Hz, 1H), 6.76 (dd, J = 24.0, 12.0 Hz, 1H), 6.70 - 6.60 (m, 1H), 6.50 - 6.32 (m, 1H), 3.74 (d, 3H) and 3.61 (d, J = 20.0 Hz, 3H).
Synthesis of (E)-1-(4-bromophenyl)-3-(4-hydroxy-3-methoxyphenyl) prop-2-en-1-one (2D)
Bromochalcone 2D was synthesized via a general procedure using vanillin as a precursor. It produces yellow powder in 95% yield. The purity is 99%, as indicated by m/z 333 in MS (EI), with a melting point of 112 °C and a boiling point of 280 °C. IR (ATR): νmax 3,361, 3,015, 2,946, 1,644, 1,572, 1,554, 1,516, 1,274, 1,028, 988, 819 and 747 cm–1. The 1H-NMR shows the proton at δ 7.85 (d, J = 10 Hz, 2H), δ 7.73 (d, J = 15 Hz, 1Hα), δ 7.61 (d, J = 10 Hz, 2H), δ 7.29 (d, J = 15 Hz, 1Hβ), δ 7.17 (d, J = 10 Hz, 1H), δ 7.10 (s, 1H), δ 6.94 (d, J = 10 Hz, 1H), δ 6.05 (s, 1H) and δ 3.94 (s, 3H).
Synthesis of (E)-1-(4-bromophenyl)-3-(4-isopropylphenyl) prop-2-en-1-one (2E)
Bromochalcone 2E was synthesized via a general procedure using cumminaldehyde as precursor. It produces white powder in 60% yield. The purity is 99%, as determined by MS (EI) at m/z 312, with m.p.: 203 °C and b.p.: -. IR (ATR): νmax 2,960, 2,925, 2,870, 1,656, 1,584, 1,509, 1,395, 1,072, 964, 825 and 736 cm–1. The 1H-NMR shows the proton at δ 7.78 (d, J = 10 Hz, 1H), δ 7.56 (d, J = 10 Hz, 1H), δ 7.41 (d, J = 10 Hz, 1H), δ 7.32 (d, J = 10 Hz, 1H), δ 7.18 (d, J = 10 Hz, 1H), δ 7.12 (d, J=15, 1Hα), δ 7.08 (d, J = 15 Hz, 1Hβ), δ 7.03 (d, J = 10 Hz, 1H), δ 6.92 (t, J = 8 Hz, 1H), δ 6.67 (d, J = 10, 1H, δ 4.07 (d, J = 10, 1H), δ 1.05 (d, J = 10, 3H) and δ 0.88 (dd, J = 3, 3H).
Synthesis of (2E,4E)-1-(4-bromophenyl)-5-phenylpenta-2,4-dien-1-one (2F)
Bromochalcone 2F was synthesized via a general procedure using cinnamaldehyde as a precursor. It produces yellow powder in 61% yield. The purity is 99%, recorded at m/z 312 in MS (EI), with m.p.: 122 °C and b.p.: 220 °C.at IR (ATR): νmax 3,026, 1,681, 1,648, 1,582, 1,008, 991, 846, 812, and 722 cm–1. The 1H-NMR shows the proton at δ 7.83 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.58 (dd, J = 16.0, 8.0 Hz, 1Hβ), 7.50 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 7.2 Hz, 1H), 7.35 – 7.31 (m, 1H) and 7.03 (d, J = 16.0 Hz, 3Hα,γ,δ).
Anticancer evaluation
Bromochalcones 2A-2F were screened for antiproliferative activity against cancer cell lines MCF-7, T47D, and HeLa, as well as the normal cell line Vero, using an MTT-based colorimetric assay. Approximately 1×104 cells per line in 100 µL were incubated in 96-well microtiter plates for 24 h. Afterward, the media were removed, and the remaining cells were treated with a serial dilution of bromochalcones 2A-2F, followed by an additional 24-hour incubation. The MTT solution was added to each well, and the incubation continued for 4 h. Additionally, 100 µL of the stopper solution, sodium dodecyl sulphate (SDS), was added to prevent formazan crystallization. After 20 h of final incubation, the wells were read at 570 nm using a microplate reader. The results were used to calculate the percentage of dead cells using Eq. (1) and the compound’s selective index using Eq. (2).

The IC50 value was determined as the antilog of y from the probit table. The percentage of cell death observed is converted to a probit value. A graph is then plotted using the equation y = ax + b, with the probit number on the y-axis and concentration on the x-axis. When y equals 5, the corresponding x value represents the Log IC50.
DFT study
Ground-state prediction was performed using Gaussian 09 with DFT calculations employing Becke’s exchange-correlation function, combining Lee, Yang, and Parr’s (B3LYP) with People’s 6- 31+g basis set. We calculate optimization of energy and frequency to obtain the highest occupied orbital energy (EHOMO), the lowest unoccupied orbital energy (ELUMO), the dipole moments (µ), and the polarizability (α). Another parameter, i.e., energy gap (ΔEH-L), chemical hardness (η), chemical softness (S), electronic affinity (EA), and ionization potential (IP) were calculated using the following equations:

Docking procedure
The proliferation proteins IGF1R (ID: 8PYL), Ki67 (ID: 1R21), and PCNA (ID: 1U7B) were downloaded from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) in .pdb format. The protein was then removed from the water molecules and the original ligand using Biovia Discovery Studio (BDS) software. However, the docking input ligands were the bromochalcones 2A-2F, which had been optimized using Gaussian 09.
Docking of 6 chalcone derivatives to 3 proliferation proteins was performed using AutoDock Vina [37]. A redocking procedure is applied for each protein with its native ligand to validate the method. The results must show the smallest RMSD value < 2. Proteins were redocked with the native ligand in a cubic grid box with dimensions 20×24×22 Å3, in a grid spacing of 1000 Å, and centered on the ligand. The best pose with the lowest RMSD was further analyzed in Pymol and BDS for visualization and interaction analysis. The s-score is recorded as the binding affinity value.
Molecular dynamics simulation
A molecular dynamics simulation was conducted to assess the stability of the protein-test compound complex. The simulation was performed using Yasara software. First, the protein location in the cell was confirmed at uniport.or.id. Since PCNA is located inside the cell, both simulations are run using the macro file md_run.mcr with modifications. The macro was prepared for MD simulation using the Amber 14 force field, with a 100 ns run at 1 bar and 310 K under a thermostat. The protein-chalcone complex is centered in a cubic box with periodic boundary conditions in all directions, with a minimum surface separation of 1.0 nm. The box contains SPC water molecules and is neutralized with Na+ and Cl- ions. The system’s energy was minimized using the steepest descent method, then equilibrated for 1 ns in an NVT ensemble, followed by 1 ns in an NPT ensemble. The Leapfrog algorithm with a time step of 2 fs is used to integrate the equations of motion. Finally, atomic coordinates were recorded every 100 ps for subsequent analysis during 100 ns simulations.
ADMET screening
Drug development involves a lengthy, multi-layered process of experimental procedures to ensure both efficacy and safety. In silico pharmacokinetic screening aims to predict potential early outcomes, enabling more effective development pathways and reducing the time and resources required for drug discovery [22,23]. For initial screening, the smile format of bromochalcones 2A-2F was uploaded to https://biosig.lab.uq.edu.au/pkcsm/ to predict Lipinski parameters. Furthermore, various parameters affecting adsorption, distribution, metabolism, and excretion were predicted for bromochalcones 2A-2F. Toxicity was assessed at pkcsm and protox3 (https://tox.charite.de/protox3/). More detailed parameters for each stage are provided in Tables S2 and S3.
Results and discussion
Synthesis progress
The 2A-2F series of bromochalcones is derived from natural aryl aldehydes. These natural products, although not yet under study, have become commodities, including vanillin, veratraldehyde, p-anisaldehyde, cuminaldehyde, and cinnamaldehyde. Exploratory natural products are costly to acquire and are scarce [24,25]. Meanwhile, natural commodity products are mass-produced with simple methods and are widely accessible [26]. They are valued not only for their stock and sustainable production, but also for their numerous biological benefits and simple structural modifications [27-29].
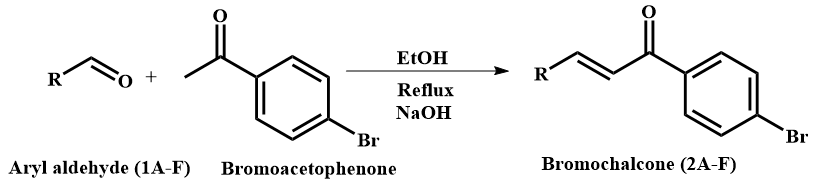
Figure 1 Aryl aldehyde precursor and the reaction scheme of bromochalcones 2A-2F.
Table 1 Summary of reaction yield.
Comp. |
Precursors |
R |
m.p. & b.p. (°C) |
Yield (%) |
Purity (%) |
2A |
Benzaldehyde |
Ph |
92; 146 |
68 |
99 |
2B |
p-Anisaldehyde |
Ph-4-OCH3 |
128; 270 |
94 |
99 |
2C |
Veratraldehyde |
Ph-3,4-(OCH3)2 |
76; 140 |
55 |
95 |
2D |
Vanillin |
Ph-3-OCH3-4-OH |
112; 280 |
95 |
99 |
2E |
Cuminaldehyde |
Ph-3-C3H7 |
203; - |
60 |
99 |
2F |
Cinnamaldehyde |
CH-Ph |
122; 220 |
61 |
99 |
In this study, chalcones were prepared by reacting aldehydes and ketones via the Claisen-Schmidt condensation (Figure 1) under basic conditions to generate the corresponding enolate, which undergoes nucleophilic addition to the aldehyde carbonyl, followed by dehydration to afford the α,β-unsaturated ketone (chalcone) framework. This substitution is followed by hydrogen rearrangement and water elimination, thereby forming an alkene. Condensation can afford E/Z (trans/cis) geometric isomers around the C=C bond, with the E (trans) isomer typically predominating under thermodynamic control. Progress was monitored by TLC using n-hexane: Ethyl acetate (1:1) as the solvent. In some chalcones, 2 spots with the same m/z were observed, indicating the presence of both trans and cis isomers. Variations in substituents at the aryl position may influence the ratio of these conformations. Benzaldehyde and cinnamaldehyde lack aryl groups, allowing the carbanion to attack from either side with equal probability, increasing the likelihood of a racemic mixture. Purification was performed by recrystallization and column chromatography. The results showed good yields (Table 1) of chalcones from para-hydroxy- and para-methoxy-substituted aldehydes (vanillin, p-anisaldehyde). In contrast, aldehydes with meta-methoxy and meta-alkyl substitutions produced smaller yields. The elucidation of the synthesis results showed typical wavenumbers of Bromochalcones, i.e., a carbonyl stretching at 1,644 - 1,681 cm– 1, a trans C-H alkene at 800 - 900 cm–1, and a C-Br band below 600 cm–1 in the FTIR spectrum (Figure S1). Their mass spectra showed molecular ions corresponding to the target molecular weight with purities of 95% - 99%. Furthermore, NMR analysis showed the presence of trans alkene protons with J values of 14-16 Hz (Figure S2).
Biological evaluation
Bromochalcones 2A-2F were evaluated using an MTT assay to assess cytotoxicity in cancer and normal cell lines. The anticancer drugs doxorubicin and cisplatin were used as a reference. The MTT assay measures mitochondrial enzyme activity in living cells [30]. Figure 2 shows that the vanillin-derived bromochalcone 2D markedly reduced cancer cell viability compared with the other analogs. The succinate dehydrogenase enzyme in mitochondria represents the number of living cells, resulting in more MTT (yellow) being converted to formazan (purple) [31]. The MTT assay is an indicator of cell growth or division. A decrease in MTT uptake indicates reduced proliferation due to sample treatment [32].
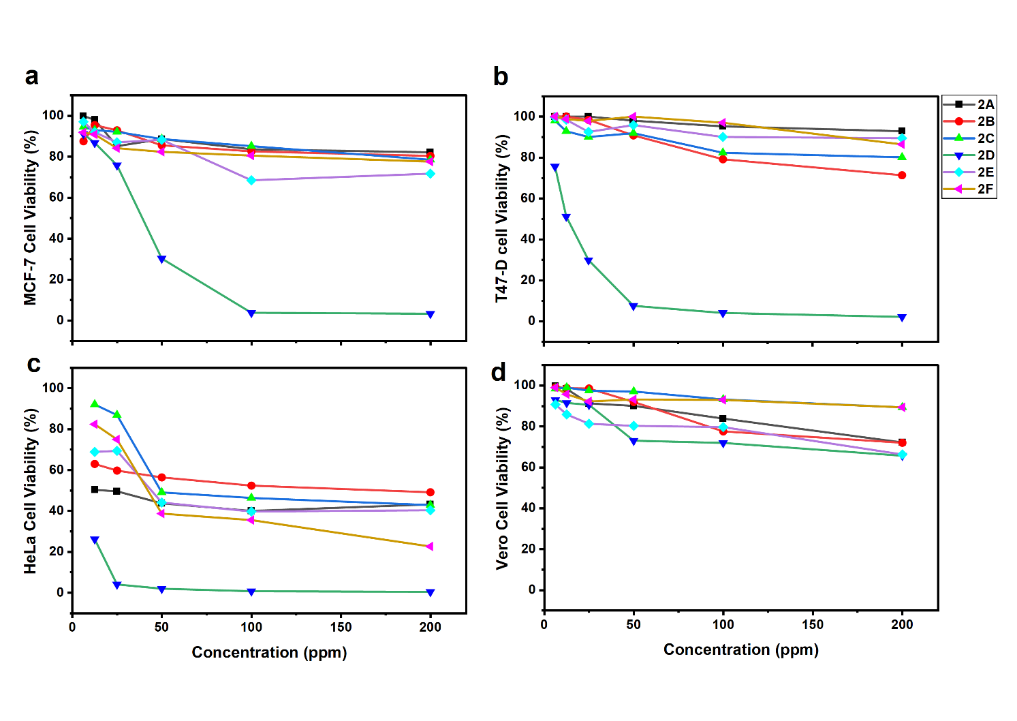
Figure 2 Cell viability of MCF-7 (a), T47-D (b), HeLa (c), and Vero (d) after 48-hour treatment with bromochalcones 2A-2F.
Bromochalcone 2B, derived from p-anisaldehyde, also inhibited T47D cell growth; however, the reduction in cell viability was not significant. Otherwise, the other bromochalcones failed to reduce cell activity or inhibit cell growth. In HeLa cells, the bromochalcone series generally reduced cell viability more than in MCF-7 and T47D, although potency varied substantially among compounds. This indicates that HeLa cells were more sensitive to bromochalcones than the other test cells. However, in normal cells, all bromochalcones exhibited minimal inhibitory activity. This pattern is desirable because it suggests preferential cytotoxicity toward cancer cells while sparing normal cells under the assay conditions.
The cell viability data were used to calculate the toxicity (IC50) and selectivity index (SI) of the candidate compounds (Table 2). The inhibition values were derived from probit analysis of the reduction in proliferative activity. Compounds with high IC50 values (> 200 µM) are considered non-toxic [33], as the IC50 must fall within a biologically relevant concentration and experimentally measurable range. The observed high IC50 in some chalcones may be attributed to insufficient test concentrations to achieve 50% lethality [34] or limited solubility in the culture medium [35]. Increasing concentration beyond the culture medium's physical limits will lead to changes in osmolarity and precipitation. Moreover, concentrations above 200 µg/mL are generally deemed therapeutically irrelevant due to inadequate potency [36].
Table 2 The calculated IC50 and selectivity index (SI) of the synthetic bromochalcones 2A-2F.
Compounds |
IC50 (µg/mL) |
|
SI |
|||||
HeLa |
MCF-7 |
T47-D |
Vero |
|
HeLa |
MCF-7 |
T47-D |
|
2A |
16 |
> 200 |
> 200 |
440 |
|
27.47 |
- |
- |
2B |
168 |
> 200 |
> 200 |
541 |
|
3.22 |
- |
- |
2C |
122 |
> 200 |
> 200 |
4,343 |
|
35.62 |
- |
- |
2D |
5 |
30 |
13 |
463 |
|
90.71 |
15.26 |
36.36 |
2E |
55 |
> 200 |
> 200 |
2,191 |
|
39.64 |
- |
- |
2F |
58 |
> 200 |
> 200 |
8,994 |
|
156.13 |
- |
- |
Doxorubicin |
- |
15 |
51 |
|
|
|
|
|
Cisplatin |
16 |
- |
- |
|
|
- |
- |
- |
Despite the insignificant decrease in viability (Figure 2), several chalcones yielded IC50 values that were too high. Therefore, they were reported only as > 200 µg/mL, indicating they were considered non-toxic. Table 2 shows that bromochalcone 2D has excellent potential as an anticancer agent for breast cancer (T47D and MCF-7) and cervical cancer (HeLa). The IC50 value even surpassed that of the standard drugs, doxorubicin in T47D and cisplatin in HeLa. These data align with the solubility data for bromochalcone 2D, which is the most soluble in water, as shown in Figure 6(a). Therefore, from the IC50 point of view, the bromochalcone derived from vanillin has the most potential as an anticancer agent.
In addition to IC50, SI data are also essential for interpreting the potential of anticancer compounds [37]. Most cytotoxic compounds not only kill cancer cells but also harm normal ones. Therefore, to differentiate the function, the selectivity index is referred to [38]. An SI value < 1 is considered more toxic to normal cells, and an SI value > 1 is considered more toxic to cancer cells [39]. Another classification of anticancer drugs is as follows: SI 1 - 3 is considered non-selective, 3 - 6 is moderately selective, and > 6 is highly selective [40]. A compound with poor selectivity usually fails at an in vivo test due to off-target toxicity [41].
Docking studies
HeLa, MCF-7, and T47D exhibit several characteristic biomarker proteins, as described in Table S1. MTT assay results indicating proliferative effects prompted molecular docking studies of the key protein that regulates this phase of cell-cycle progression. PCNA was selected because of its central role in DNA replication and repair in proliferating cells, including HeLa, MCF-7, and T47D. IGF1R was included because it was overexpressed and sensitive in MCF-7, while Ki67 served as a general proliferation marker. IGF1R levels in MCF-7 are expressed more and are more sensitive than in T47D [42-44]. Meanwhile, PCNA levels in T47D are 4-6 times higher than those in MCF-7 [45].
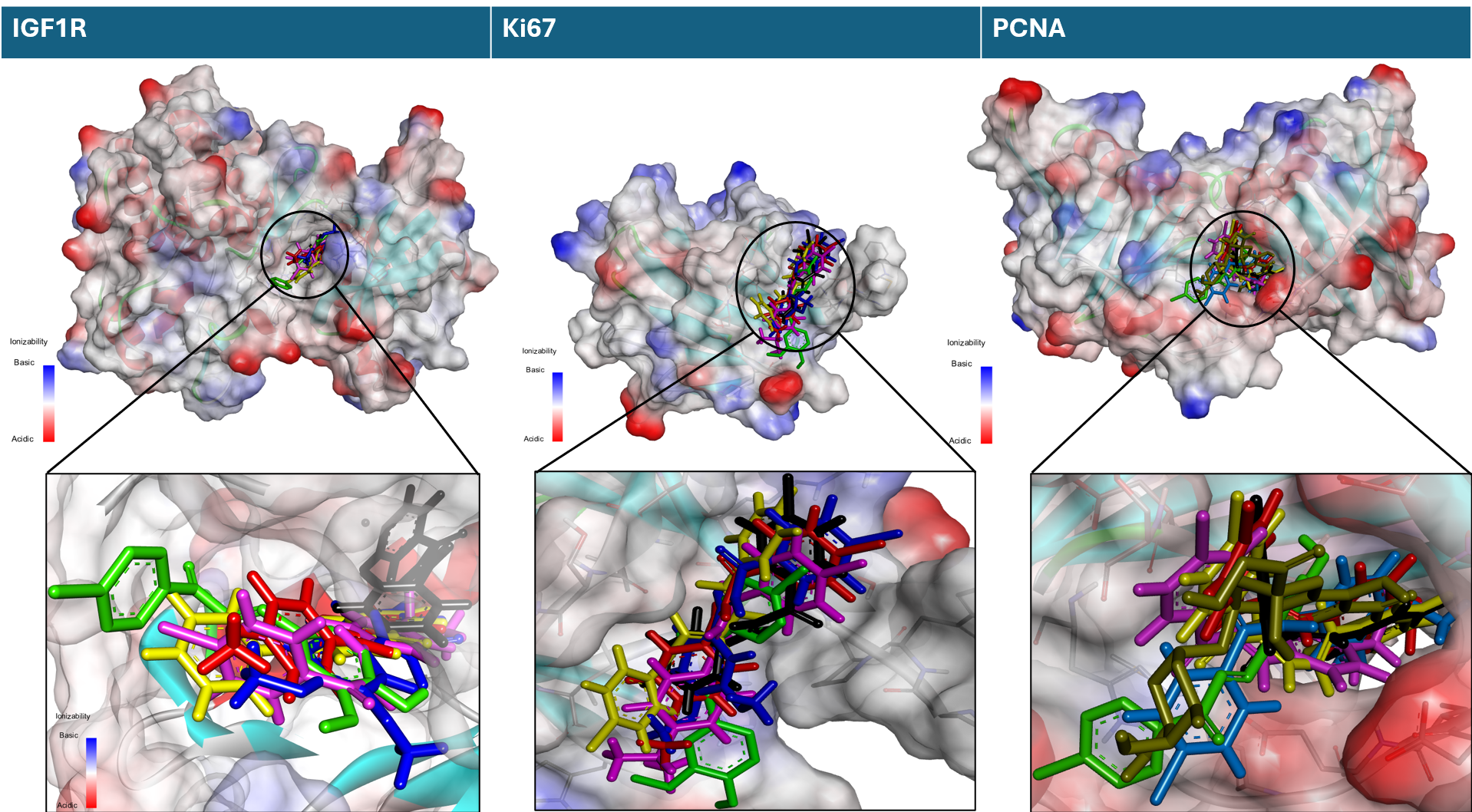
Figure 3 Superimposed bromochalcones on the binding pocket proteins IGF1R, Ki67, and PCNA (black 2A, red 2B, blue 2C, green 2D, pink 2E, and yellow 2F).
The correlation between docking scores and observed in vitro potency was examined to identify which target best aligned with the observed activity trend, while acknowledging that docking provides only a preliminary indication of target engagement. Hence, docking studies were performed on PCNA, IGF1R, and Ki67 to estimate the potential interactions of bromochalcones 2A-2F with these proliferation-related proteins. Visualization of the chalcone interaction in the pocket binding site of each protein is presented in Figure 3. The interaction’s effectiveness was evaluated using the binding affinity parameter (Table 3), which was subsequently validated against the in vitro IC50 value.
Table 3 Binding affinity and inhibition constant of bromochalcones 2A-2F.
Compounds |
Binding
Affinity |
|
Inhibition
Constant |
IC50 |
||||
IGF1R |
Ki67 |
PCNA |
|
IGF1R |
Ki67 |
PCNA |
||
2A |
–7.3 |
–5.5 |
–5.9 |
|
0.44 |
9.24 |
4.70 |
16 |
2B |
–7.5 |
–5.6 |
–5.4 |
|
0.31 |
7.81 |
10.95 |
168 |
2C |
–7.9 |
–5.8 |
–5.4 |
|
0.16 |
5.57 |
10.95 |
122 |
2D |
–7.5 |
–5.7 |
–6.2 |
|
0.31 |
6.59 |
2.83 |
5 |
2E |
–8.1 |
–6.2 |
–6.1 |
|
0.11 |
2.83 |
3.35 |
55 |
2F |
–8.0 |
–5.9 |
–5.8 |
|
0.13 |
4.70 |
5.57 |
58 |
IE0 |
–7.1 |
- |
- |
|
|
|
|
- |
Correlation |
–0.62 |
–0.36 |
0.84 |
|
–0.63 |
–0.43 |
0.88 |
|
Binding affinity (docking score) is commonly used to rank predicted ligand-protein interactions. It highlights the strength of the ligand-protein interaction, with a more negative binding affinity indicating a stronger, more stable interaction. Table 3 shows the different binding affinities of chalcones for each protein. The ΔG values for IGF1R and Ki67 proteins are 2E < 2F < 2C < 2D, while 2B < 2A. In contrast, for PCNA proteins, the values are 2D < 2E < 2A < 2F < 2C < 2B. Aligned with the IC50 values from in vitro studies, the most similar pattern is observed in chalcone-PCNA binding affinity. Since the docking results for IGF1R and Ki67 did not match the in vitro pattern, further analysis focused on the interaction between bromochalcones 2A-2F and PCNA.
Table 4 PCNA-ligand interactions.
Compounds |
ΔG
|
Essential interactions |
||
H-bond (Å) |
Hydrophobic |
Other |
||
2A |
–5.9 |
His44 (1.95) |
Pi-sigma Val123 (3.82), pi-alkyl Leu121 (5.30) |
|
2B |
–5.4 |
His44
(2.51) |
Alkyl Cys27, Alkyl Val123, Pi-Alkyl Leu121 |
|
2C |
–5.4 |
Cys27 (2.67) Glu124 (1.99) |
Alkyl Cys27, Alkyl Leu121, Alkyl Val123 |
|
2D |
–6.2 |
Glu25 (2.80) Met40 (2.95) Met40 (2.49) |
π-π stacked His44 (3.67) |
|
2E |
–6.1 |
His44 (2.79) |
|
Pi-anion Glu124 (4.06) |
2F |
–5.8 |
His44 (2.39) |
Pi-sigma Val123 (3.87), pi-Alkyl Leu121 (5.27) |
|
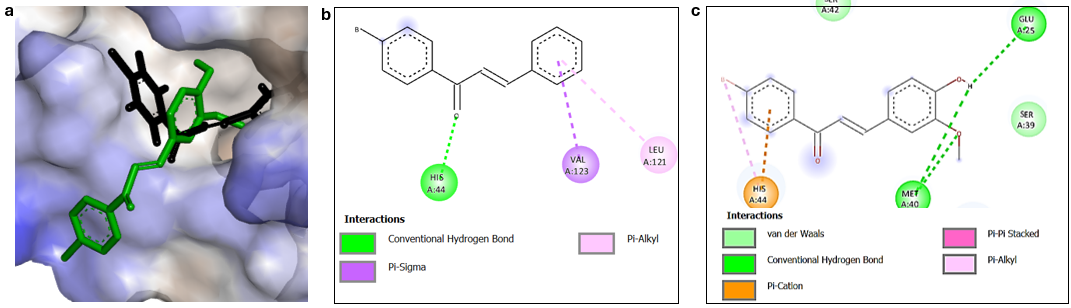
Figure 4 Hydrofobic surface map (a), and 2-dimensional interaction of bromochalcones 2A (b) and 2D (c) in the PCNA binding pocket.
The best chalcone-PCNA interaction was achieved by the bromochalcone 2D, yielding a ΔG value of –6.2 kcal/mol and a Ki value of 2.83×10−5 µmol. Molecular docking of this complex revealed 3 hydrogen bond interactions with distances of 2.49, 2.80, and 2.95 Å involving the amino acid residues Met40 and Glu25 (Table 3). The side chains of the OH and OCH3 groups in the vanillin precursors have the most significant impact on this interaction. In addition, the phenyl ring of the bromoacetophenone fragment is predicted to engage in π–π stacking and π–cation interactions with His44. These interactions facilitate bromochalcone 2D’s entry into the active-site pocket (Figure 4). Compared to other ligands, bromochalcone 2D fits most appropriately in this pocket, while other ligands tend to be slightly outside it. Docking shows that our compound can bind to the protein, but we need to examine the interaction dynamics over time in the next chapter.
Molecular dynamics simulation
MD simulations were performed to assess the interaction stability, flexibility, and reliability of the protein-ligand complex. The previous chapter identified PCNA as the preferred protein because of its strong correlation between ΔG values from molecular docking and in vitro IC50 values. PCNA is a ring protein (Figure 5(a)) that consists of 3 monotrimers (Figure 5(b)), an essential protein in cell biology that functions as a DNA “clamp” to assist replication and repair, allowing DNA polymerase to divide cells efficiently. Two ligands with the best IC50 values were subjected to molecular dynamics simulations. This simulation was performed using the Amber 14 force field in YASARA for 100 ns [46].
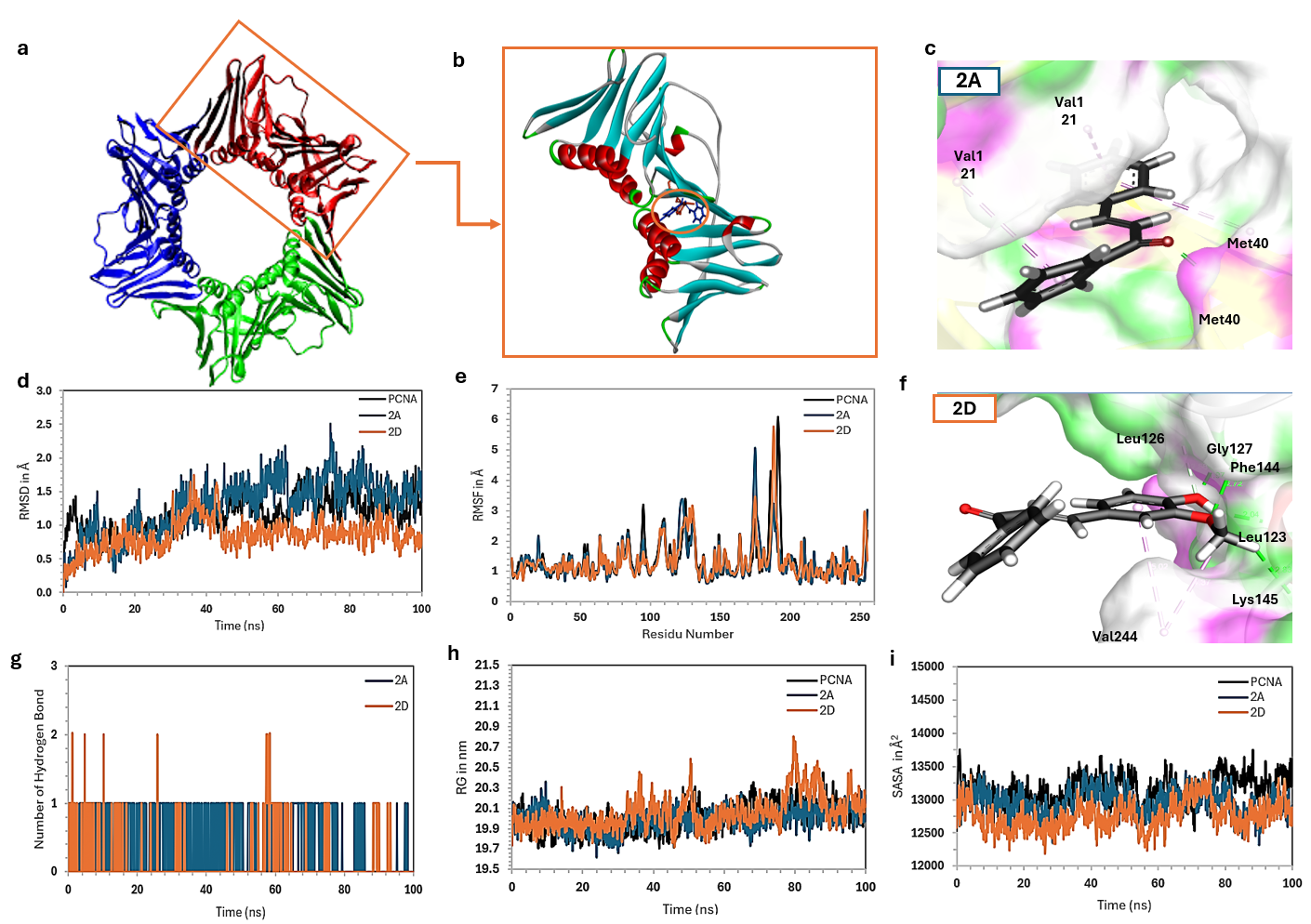
Figure 5 Compilation of PCNA molecule structure (a), monotrimer PCA-ligand complex (b), interactions of bromochalcone 2A (c), and bromochalcone 2D (f) in the binding site, RMSD (d), RMSF (e), number of H-bonds (g), Rg (h), and SASA (i) of bromochalcones 2A (green) and 2D (orange) with PCNA.
The overall stability of the system was evaluated by monitoring the RMSD values of the protein-ligand complex (Figure 5(d)) [47]. The simulation reached equilibrium after running for 50 ns. The RMSD fluctuated between 0.871 and 2.504 Å for bromochalcone 2A, while bromochalcone 2D fluctuated between 0.521 and 0.882 Å. The average RMSD was 1.561 and 0.882 Å for bromochalcones 2A and 2D, respectively. These values are reasonable for a protein-ligand system and comparable to the alo-PCNA (0.827 - 1.871 Å, average 1.310 Å) [48]. The relatively stable RMSD profiles suggest that ligand binding does not induce significant conformational changes in the overall PCNA structure. The binding of bromochalcones 2A and 2D is expected to act as inhibitors at the surface pocket of the interdomain connecting loop (IDCL) [49]. This also explains that bromochalcone 2A is sufficient for initial binding and stabilization. Still, the addition of para-hydroxy and meta-methoxy groups to ring B (bromochalcone 2D) provides additional contact points, thereby optimizing the complex's interactions and stability.
The IDCL amino acid residues in PCNA are located between 120 and 135. The dynamics of this movement are evident in RMSF (Figure 5(e)). Bromochalcone 2A lessens IDCL fluctuations, making the changes more stable, while 2D takes it a step further by reducing them even further. Bromochalcone 2A relies on the hydrophobic bonds of Val123 and Leu121, while bromochalcone 2D relies on a true hydrogen bond with Gly127 and a carbon H-bond with Leu126 (Figure 5(c)). Both compounds bind to the IDCL site, consistent with greater stabilization of local flexibility in the bromochalcone 2D complex. In addition, bromochalcone 2D forms another H-bond at residue Phe144, whereas the only H-bond by bromochalcone 2A appears to interact with Met40 (Figure 5(f)). Outside the main IDCL region, RMSF also decreases at residues 50 - 55, 85 - 95, 120 - 135, and 180 - 195. As a result of these interactions, bromochalcones 2A and 2D stabilize the movement of the gating loop for DNA, the subunit interface, and the C-terminal tail [49]. It’s like an allosteric key that locks the stiffness of the other 3 dynamic regions of PCNA [50].
The radius of gyration (Rg) is a measure of the global compactness of a protein/protein complex. PCNA shows a gentle fluctuation of about 0.6 Å, ranging from 19.8 to 20.4 Å. Meanwhile, the complex remains smooth around 19.6-20.3 Å, with bromochalcones 2A and 2D fluctuating slightly more, between 19.7 and 20.7 Å, respectively. This is normal for protein with a ~29 kDa mass. The decrease in Rg in the complex indicates that binding of bromochalcones 2A and 2D causes PCNA to adopt a more complex conformation, consistent with the RMSF, which shows stabilization in several critical loops, including IDCL, the gating loop, the subunit interface, and the tail. This stability reduces the tendency of atoms to move away from the protein’s center of mass. When bromochalcone 2A and PCNA show a highly correlated pattern, bromochalcone 2D exhibits a slight increase at 50 and 80 ns (Figure 5(h)), considering that 2D is a more potent inhibitor than 2A (in vitro), this fluctuation may not indicate instability. Instead, it is a more complex allosteric dynamic. We suspect that the polar interaction of bromochalcone 2D motion suggests that the best inhibitors are often not the most stabilizing, but rather the most effective at disrupting the protein’s natural dynamics toward the active state [51].
SASA captures the protein’s natural dynamics, with water molecules serving as solvents. Complexes 2A and 2D show a very similar pattern to Complex alo, and importantly, both remain consistent under PCNA fluctuations (Figure 5(i)). The decrease in exposed solvent area results from ligand binding in the pocket IDCL, which covers the surface area previously exposed to solvent. Reduced SASA fluctuations may also suggest decreased surface breathing in the ligand-bound complexes. Although they exhibit similar macroscopic dynamics, bromochalcones 2A and 2D differ in their potential strengths. The bonding details clearly influence this difference due to the presence of additional groups in the aldehyde source [46].
DFT Study
DFT is used to understand the electronic properties and molecular structure, as well as to predict the reactivity of drug compounds [52-55]. The optimal structure significantly influences binding affinity to biological targets, such as enzymes and other proteins. It is also used as input in molecular docking simulations. The calculated DFT parameters in this study include HOMO energy (EHOMO), LUMO energy (ELUMO), HOMO-LUMO energy gap (ΔEH-L), dipole moment (µ), polarizability (α), hardness (η), and softness (s).
Figure 6 illustrates the frontier molecular orbital (LUMO and HOMO) density of the optimized investigated molecule. EH represents the energy required to provide an electron in its orbital for interaction. At the same time, EL indicates the amount of energy required to accept electrons during interactions with other molecules or the environment. The interaction between these 2 orbitals of a molecule determines the structure of the transition state. Examining the relationship between EHOMO, ELUMO, and ΔEH-L with docking-derived ΔG values shows that ΔEH-L is uncorrelated with ΔG. At the same time, EH negatively correlates, and EL positively correlates—though less consistently. These findings suggest that electrostatic forces, rather than charge transfer, primarily govern the binding mechanism. The active site of the protein contains numerous negatively charged regions; consequently, ligands with high EH (strong donors) may experience repulsion due to electronic effects. The main factors influencing interaction strength are hydrogen bonds, van der Waals forces, and controlled electronic interactions. In this case, steric factors and shape complementarity play a more crucial role than electronic reactivity in determining binding affinity. Consequently, the ΔEH-L parameter is relatively weak.
Another parameter of the investigated compounds is presented in Table 5. The dipole moment of bromochalcones 2A-2F is positively correlated with the ΔG value. As the dipole moment increases, the binding affinity also increases. Bromochalcones 2D and 2E, with dipole moment values greater than 4.6, achieve the strongest ΔG values, while bromochalcones 2A-2C, with dipole moments less than 4.6 D, are the weakest. Polarizability is positively correlated with ΔG, but the effect is weaker (2F, with the highest polarizability, does not have the strongest interaction). Dipole moment influences solubility and membrane permeability. A higher dipole moment makes drugs more polar, increasing water solubility and suitability for water-soluble transport [56]. However, as biological membranes are hydrophobic, highly polar drugs may have lower permeability. Drugs with an optimal dipole moment can distribute effectively in biological environments. Polarizability is also crucial, as it enhances van der Waals interactions, thereby improving binding stability to targets. Greater polarizability is usually associated with higher lipophilicity, which enhances membrane permeability, pharmacological selectivity, and bioavailability, while reducing off-target effects [57].
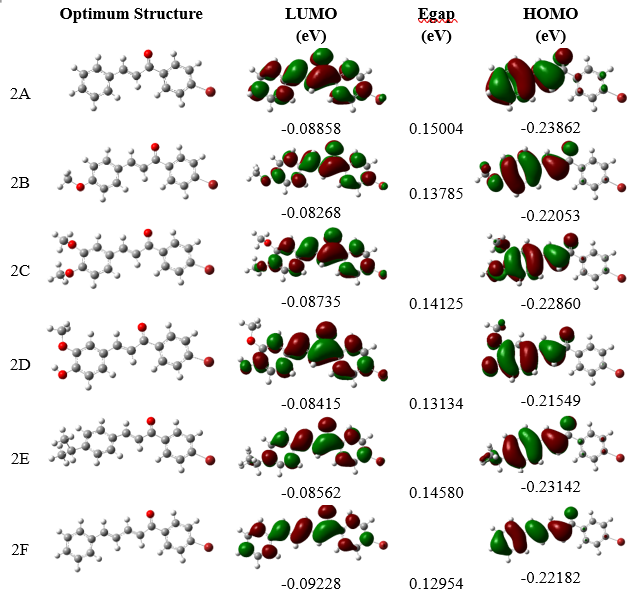
Figure 6 Frontier LUMO and HOMO orbitals of bromochalcones 2A-2F.
Table 5 Quantum chemical characteristics of the investigated molecules in vacuum.
Compounds |
ELUMO
|
EHOMO
|
ΔE
H-L |
µ
|
α
|
Hardness (η) |
Softness (s) |
2A |
−0.0885 |
−0.23862 |
0.15004 |
3.89075 |
192.819 |
0.07502 |
13.32978 |
2B |
−0.0841 |
−0.21549 |
0.13134 |
3.73834 |
225.406 |
0.06567 |
15.22765 |
2C |
−0.0873 |
−0.2286 |
0.14125 |
4.37593 |
235.212 |
0.07063 |
14.15929 |
2D |
−0.0856 |
−0.23142 |
0.14580 |
4.63237 |
235.963 |
0.07290 |
13.71742 |
2E |
−0.0826 |
−0.22053 |
0.13785 |
5.11375 |
222.036 |
0.06893 |
14.50852 |
2F |
−0.0922 |
−0.22182 |
0.12954 |
4.65696 |
249.966 |
0.06477 |
15.43925 |
Correlation |
−0.1180 |
0.3460 |
−0.4128 |
0.6045 |
0.1205 |
−0.4127 |
0.4118 |
The hardness and softness of the ligand also affect binding affinity and specificity [58]. Soft ligands tend to bind to soft sites in proteins, leading to covalent bonding, electron transfer, and dipole interactions. The flexibility of their electron cloud optimized binding. In contrast, hard ligands prefer to interact with hard sites, forming electrostatic or hydrogen bonds, which results in less charge transfer [59]. Hydrogen bonds are relatively strong and exhibit low binding affinity [60]. DFT calculations show a positive correlation between softness and ΔG, while the opposite occurs for hardness and ΔG. Notably, compounds at the extremes of hardness/softness did not necessarily yield the most favorable docking scores, suggesting that balanced electronic properties may be advantageous. Ligands that are too hard are less reactive, and those that are too soft are less selective. Bromochalcones 2D and 2E are in equilibrium between stability and reactivity. Based on this data, further drug design aims to increase the dipole moment to more than 4.5 D, keep hardness within the range of 0.068 - 0.073, target an energy gap between 0.138 - 0.145 eV, and note that polarizability is not critical if it exceeds 220.
Drug likeness and ADMET
Drug likeness and ADMET properties were used to screen these chalcones for further development as drug candidates. Drug-likeness is an essential feature that underpins the selection of effective therapeutic candidate compounds. Table 6 displays properties of potential drugs, bromochalcones 2A-2F, from pkcsm. The Lipinski method evaluates the drug-likeness of these potential compounds. It relies on Lipinski’s rule of 5, which includes 5 criteria: (1) molecular weight under 500 g/mol, (2) lipophilicity or partition coefficient (mLogP) below 5, (3) TPSA under 140 Ų, (4) fewer than 5 HBD, and (5) fewer than 10 HBA. Molecules meeting at least 3 of these criteria are likely to exhibit good absorption (Lipinski, 2004). The majority of synthesized chalcones comply with Lipinski’s rule, as they possess an approximate molecular weight of 300 g/mol, with hydrogen bond acceptors (HBA) and donors (HBD) below 5, and a topological polar surface area (TPSA) below 140 Ų. Their average logP (mLogP) is approximately 4.3, providing an optimal balance between maintaining cell membrane permeability and facilitating transport into the bloodstream for systemic distribution. Nonetheless, the conjugation of a chalcone chain at position bromochalcone 2F and an isopropyl side chain at position bromochalcone 2E increases their lipophilicity. Notably, the bromochalcone 2E surpasses the limits set by Lipinski’s rule, rendering it somewhat less suitable as a drug candidate due to potential distribution challenges. Overall, in accordance with Lipinski’s rule, an additional 5 chalcones demonstrate potential as therapeutic agents, comparable to the natural product aloe-emodin and surpassing doxorubicin.
Table 6 The molecular descriptor of bromochalcones 2A-2F obtained from pkcsm.
Compounds |
MW |
mLogP |
R-bonds |
nHBA |
nHBD |
TPSA |
Violation |
2A |
287.156 |
4.3452 |
3 |
1 |
0 |
108.923 |
0 |
2B |
317.182 |
4.3538 |
4 |
2 |
0 |
120.401 |
0 |
2C |
347.208 |
4.3624 |
5 |
3 |
0 |
131.880 |
0 |
2D |
333.181 |
4.0594 |
4 |
3 |
1 |
125.196 |
0 |
2E |
329.237 |
5.4686 |
4 |
1 |
0 |
128.018 |
1 |
2F |
313.194 |
4.9014 |
4 |
1 |
0 |
120.963 |
0 |
Aloe-emodine |
254.241 |
1.6599 |
1 |
4 |
2 |
108.489 |
0 |
Doxorubicin |
543.525 |
0.0013 |
5 |
12 |
6 |
222.081 |
3 |
Absorption and distribution data of bromochalcones 2A-2F are provided in Table S2. They showed predicted water solubility values of approximately −4 to −5 (log mol/L). In contrast, the standard drug doxorubicin and Aloe-emodine, a potential natural product for anticancer applications, have solubilities of approximately 3 mol/L. This suggests that bromochalcones 2A-2F are predicted to be less water-soluble than doxorubicin, which may limit oral bioavailability because absorption requires adequate dissolution in gastrointestinal fluids. Low water solubility also reduces intestinal absorption; however, these data indicate that intestinal absorption of chalcone is significant. This is supported by good permeability values (> 1; log Papp in 10 - 6 cm/s) for bromochalcones 2A, 2D-2F.
Since bromochalcones 2A-2F have poor water solubility but still meet Lipinski’s rule, the P-glycoprotein transporter mechanism may support their distribution. There may be a potential need to develop prodrug strategies, nanoparticles, or solubilizing excipients. Coupling this data in Figure 7(a) shows that doxorubicin and aloe-emodine, as comparable standards, have good solubility but very low permeability. Such properties may partially explain why doxorubicin is commonly administered via non-oral routes in clinical practice. Of the 6 chalcones, bromochalcones 2B and 2C are included in the high-risk category. Bromochalcone 2A has moderate permeability but poor solubility. Bromochalcone 2F has excellent permeability, but low solubility. It requires formulation improvement. Bromochalcone 2D has the highest permeability and the lowest solubility. Therefore, bromochalcone 2D from vanillin is the best choice for adsorption.
Distribution parameters (Figure 7(b)) describe how the drug moves after absorption, affecting dosage, efficacy, and safety. The volume of distribution (Vd) and steady-state volume of distribution (VDS) determine the drug’s distribution in tissues. The ideal range for VDSs is 0.3 - 1, while for chalcone, it is −0.017 to 0.623, with the highest value observed for bromochalcone 2E. Higher VDSs indicate greater tissue penetration of the drug. Fraction unbound (Fu) data show very high values except for Aloe. Ultra-high Fu may limit free-drug availability, leading to increased doses or to switching to different formulations to maintain efficacy. BBB penetration > 0.3 CNS indicates very high penetration in the brain, while a BBB with a negative value indicates that the drug stays out of the brain. A low BBB value may help avoid neurotoxic side effects in peripheral targets.
The metabolic profile of bromochalcones 2A-2F on cytochrome P450 in Figure 7(c) shows that all bromochalcones inhibit CYP1A2/2c9. Therefore, this drug candidate should be considered for concomitant use with caffeine and clopidogrel. The excretion profile (Figure 7(d)) shows high doxorubicin clearance, consistent with its short half-life. Meanwhile, almost all chalcones are negative (except 2C), meaning they circulate systemically longer and have longer half-lives. Therefore, judicious dosing is required to avoid cumulative toxicity. Furthermore, bromochalcone 2B (red) is the only chalcone to show positive interaction at the OCT2 substrate, thereby increasing the risk of nephrotoxicity. Encouraging for cardiac safety, improving upon doxorubicin, which is widely recognized as cardiotoxic [61]. For AMES and genotoxicity assays, Aloe-emodin, a natural product used as a standard of comparison, yields a positive result. Coupled with its high Fu value, it may cause genotoxicity through DNA interaction. Skin sensitization data indicate that bromochalcones 2A, 2E, and 2F are positive, although their skin permeability (Figure 7(a)) is low (log kp < 1.97), which reduces the risk of skin sensitization. Bromochalcones 2A, 2E, and 2F can still be considered, but they are classified as moderately toxic. Bromochalcone 2C was found to pose a risk of hepatotoxicity, despite being administered at a high tolerated dose (1.374 mg/kg/day) (Table S4). However, its low clearance (−0.148 log mL/min/kg) will allow it to continue circulating, bypassing the liver. Plus, its interaction with CYP3A4i increases its risk to the liver.
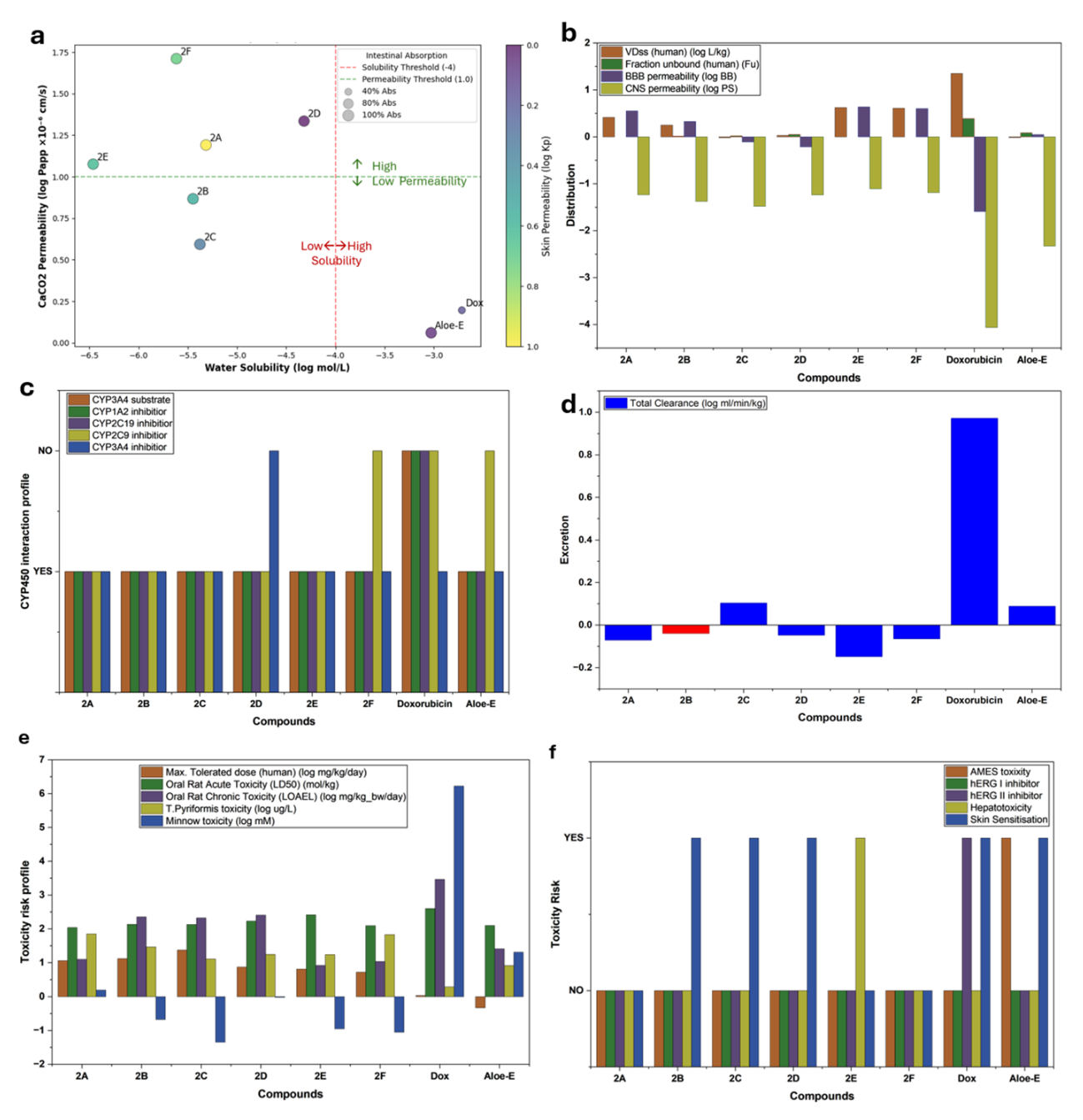
Figure 7 Compilation data of adsorption (a), distribution (b), metabolism (c), excretion (d), and toxicity (e,f) properties of bromochalcones 2A-2F, comparable with doxorubicin and aloe-emodin.
The toxicity profile (Figures 7(e) and 7(f)) indicates that all chalcones are hERG-negative. This ADMET study concluded that doxorubicin is limited by its high cardiotoxicity. Aloe-emodin has not yet been developed and poses a genotoxic risk. Chalcones offer advantages over doxorubicin, including a better safety profile, no hERG-mediated cardiotoxic interactions, a higher tolerated dose, and fewer drug interactions. These chalcones have potential as drugs, with the priority and development stages listed in Table 7.
Table 7 ADMET prediction priority.
Rank |
Comp. |
Key strength |
Weakness |
Development |
1 |
2C |
Highest MTD, no toxicity |
Low solubility |
Oral formulation optimization |
2 |
2F |
Best permeability |
Skin sensitization |
High-bioavailability oral drugs |
3 |
2D |
Cyp3A4 inhibition |
Drug interaction risk |
Combination therapies |
4 |
2A |
Balance ADME |
Skin sensitization |
IV/oral with enhancers |
5 |
2B |
Cardiac safety |
OCT2 nephrotoxicity |
Non-renal indication |
6 |
2E |
Tissue penetration (VDss) |
Hepatotoxicity |
Reformulation required |
Although bromochalcone 2C has the best ADMET potential, its low solubility makes it challenging to form an oral preparation. This property may also explain the low in vitro IC50 values. Bromochalcone 2F, as the next-best candidate, failed to achieve a good IC50. The low IC50 value is attributed to the compound’s molecular structure, which does not facilitate robust interactions with the proliferation protein PCNA. Bromochalcone 2D, with the lowest IC50, is also the third best in ADMET prediction. In addition, bromochalcone 2D was shown to interact firmly and stably with PCNA in molecular docking and molecular dynamics simulations. Bromochalcone 2A is a potential drug candidate based on ADMET after bromochalcone 2D. It also exhibits an IC50 value in the strong category, and its binding affinity for PCNA is relatively strong. Bromochalcone 2A is the second candidate recommended for further research after bromochalcone 2D. Despite weaknesses in skin sensitization, 2A could be used as a drug with intravenous preparations. Bromochalcone 2B did not achieve an outstanding IC50; it fell into a weak category. Docking, molecular dynamics, and DFT simulations showed that bromochalcone 2B does not interact very strongly with PCNA. The best news is that bromochalcone 2B is cardiac-safe, with the caveat of nephrotoxicity; it may still be developed for patients with non-renal indications. Finally, bromochalcone 2E with a moderate IC50 and a high mlogP value makes it unacceptable as a drug candidate requirement. High mlogP facilitates tissue penetration; however, its distribution through the bloodstream is challenging. Bromochalcone 2E is also dangerous for the liver. In conclusion, among the factors discussed in this chapter, bromochalcones 2D and 2A are the best, with IC50 values comparable to those of doxorubicin. Bromochalcones 2D and 2A warrant continued evaluation as anti-cervical cancer candidates in HeLa cells, with a focus on inhibiting proliferation.
Conclusions
Following the synthesis and evaluation of bromochalcones derived from natural aryl aldehydes using in vitro and in silico approaches, our findings indicate that bromochalcone 2D (vanillin-derived) exhibited the most favorable antiproliferative profile among the tested series. In vitro screening using the MTT assay yielded an IC50 value surpassing that of doxorubicin. Molecular docking analysis targeting 3 principal proliferation-associated proteins (IGF1R, Ki67, and PCNA) revealed that the ΔG values of all proposed chalcones with PCNA showed the highest correlation with the in vitro IC50, exceeding 80%. Molecular dynamics simulations confirmed the stability of hydrogen-bond interactions within the 2D-PCNA complex. Density Functional Theory (DFT) calculations estimated the dipole moment and softness of the compounds, which support robust hydrogen-bonding and pi-cation interactions with this protein. Ultimately, drug-likeness and ADMET assessments indicated that most chalcone samples are viable candidates for further drug development, except for 2E and 2F, which exhibited excessively high mlogP values, potentially impairing distribution and increasing the likelihood of off-target effects due to their ease of permeability across various tissues and cells. Future design efforts aim to enhance the dipole moment to values exceeding 4.5 D, maintain hardness within 0.068 - 0.073, target an energy gap of 0.138 - 0.145 eV, and keep polarizability below 220.
Acknowledgements
The author gratefully acknowledges the Austria-Indonesia Centre (AIC) for computational chemistry, which provided access to and licenses for Gaussian 09. We are also grateful to the Indonesian Education Scholarship (BPI), the Center for Higher Education Funding and Assessment (PPAPT), the Ministry of Higher Education, Science, and Technology of the Republic of Indonesia, and the Indonesia Endowment Fund for Education (LPDP) for their support of this research.
Declaration of Generative AI in Scientific Writing
All scientific content, analysis, interpretation, and conclusions presented in the paper were developed independently by the authors.
CRediT Author Statement
Amri Setyawati: writing-original draft, methodology, investigation, formal analysis. Endang Astuti: writing-review, editing, supervision in anticancer. Niko Prasetyo: writing-review, editing, supervision in silico. Indriana Kartini: writing-review, editing, supervision in elucidation. Tutik Dwi Wahyuningsih: writing-review, editing, conceptualization, supervision in synthesis.
References
[1] F Bray, M Laversanne, H Sung, J Ferlay, RL Siegel, I Soerjomataram and A Jemal. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 2024; 74(3), 229-263.
[2] JR Wei, MY Lu, TH Wei, JS Fleishman, H Yu, XL Chen, XT Kong, SL Sun, NG Li, Y Yang and HW Ni. Overcoming cancer therapy resistance: From drug innovation to therapeutics. Drug Resistance Updates 2025; 81, 101229.
[3] ZA Mahdi, BSA Almjalawi, M Naimuzzaman, M Hasan, FT Al-Alaq, HOM Al-Dahmoshi, A Kumer, NSK Al-Khafaji, SAM Al-Yasiri and M Saki. Exploring camptothecin derivatives from the Chinese tree of Camptotheca acuminata as breast cancer protease inhibitors: Insights from multi-scale computational analysis. The Open Bioinformatics Journal 2025; 18(1), e18750362369549.
[4] T Nakaphan, M Teerachaisakul, K Sawanyawisuth and K Pongpirul. Herbal product availability and consumption patterns: A comprehensive scoping review. Natural Resources for Human Health 2025; 5(4), 522-531.
[5] J Tauchen, L Huml, M Jurášek, JM Regenstein and F Ozogul. Synthetic and semi-synthetic antioxidants in medicine and food industry: A review. Frontiers in Pharmacology 2025; 16, 1599816.
[6] B Goel and SK Jain. Semisynthesis: Bridging natural products and novel anticancer therapies. European Journal of Medicinal Chemistry Reports 2024; 12, 100218.
[7] D Pereira, RT Lima, A Palmeira, H Seca, J Soares, S Gomes, L Raimundo, C Maciel, M Pinto, E Sousa, M Helena Vasconcelos, L Saraiva and H Cidade. Design and synthesis of new inhibitors of p53-MDM2 interaction with a chalcone scaffold. Arabian Journal of Chemistry 2019; 12(8), 4150-4161.
[8] S Safe. Natural products as anticancer agents and enhancing their efficacy by a mechanism-based precision approach. Exploration of Drug Science 2024; 2(4), 408-427.
[9] K Ganesan, C Xu, B Du, J Che, F Gao, C Zheng and J Chen. Isoliquiritigenin: A natural compound with a promising role in inhibiting breast cancer lung metastasis. Journal of Traditional and Complementary Medicine 2024; 15(7), 702-713.
[10] R Palanisamy, NI Kahingalage, D Archibald, I Casari and M Falasca. Synergistic anticancer activity of plumbagin and xanthohumol combination on pancreatic cancer models. International Journal of Molecular Sciences 2024; 25(4), 2340.
[11] K Okoniewska, MT Konieczny, K Lemke and T Grabowski. Pharmacokinetic studies of oxathio-heterocycle fused chalcones. European Journal of Drug Metabolism and Pharmacokinetics 2017; 42(1), 49-58.
[12] E Winter, C Dal Pizzol, C Locatelli, AH Silva, A Conte, LD Chiaradia-Delatorre, RJ Nunes, RA Yunes and TB Creckzynski-Pasa. In vitro and in vivo effects of free and chalcones-loaded nanoemulsions: Insights and challenges in targeted cancer chemotherapies. International Journal of Environmental Research and Public Health 2014; 11(10), 10016-10035.
[13] S Chowdhary, Preeti, Shekhar, N Gupta, R Kumar and V Kumar. Advances in chalcone-based anticancer therapy: Mechanisms, preclinical advances, and future perspectives. Expert Opinion on Drug Discovery 2024; 19(12), 1417-1437.
[14] R Hassan, S Emam, D Hwang, G Do Kim, S Hassanin, M Khalil, A Abdou and A Sonousi. Design, synthesis and evaluation of anticancer activity of new pyrazoline derivatives by down-regulation of VEGF: Molecular docking and apoptosis inducing activity. Bioorganic Chemistry 2022; 118, 105487.
[15] RA Sheldon, ML Bode and SG Akakios. Metrics of green chemistry: Waste minimization. Current Opinion in Green and Sustainable Chemistry 2022; 33, 100569.
[16] A Kumar, G Soumya, V Kanchana, K Singh, N Maurya, S Kumar, A Singh, J Kumar, K Srinivas, D Chanda, S Luqman, S Misra, A Meena and B Balakishan. Design, synthesis, and in silico docking studies of novel cinnamaldehyde-chalcone derivatives with anti-cancer potential and in vivo acute oral toxicity profiling. RSC Advances 2025; 15(37), 30627-30638.
[17] HH Wang, KM Qiu, HE Cui, YS Yang, M Xing, XY Qiu, LF Bai and HL Zhu. Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives containing benzodioxole as potential anticancer agents. Bioorganic & Medicinal Chemistry 2013; 21(2), 448-455.
[18] M Saifullah, A Hasan, M Kaleem, E Raneem, A Gupta, M Amir, M Mumtaz Alam, M Akhter, S Tasneem and M Shaquiquzzaman. A comprehensive review of structure activity relationships: Exploration of chalcone derivatives as anticancer agents, target-based and cell line-specific insights. Medicine in Drug Discovery 2025; 28, 100230.
[19] R Michalkova, M Kello, M Cizmarikova, A Bardelcikova, L Mirossay and J Mojzis. Chalcones and gastrointestinal cancers: Experimental evidence. International Journal of Molecular Sciences 2023; 24(6), 5964.
[20] C Gracesella, H Patintingan and M Louisa. Anticancer potential of chalcone and its derivatives against triple negative breast cancer. Journal of Chemical Health Risks 2024; 14(3), 309-316.
[21] L Gu, R Lingeman, F Yakushijin, E Sun, Q Cui, J Chao, W Hu, H Li, R Hickey, J Stark, Y Yuan, Y Chen, S Vonderfecht, T Synold, Y Shi, K Reckamp, D Horne and L Malkas. The anticancer activity of a first-in-class small-molecule targeting PCNA. Clinical Cancer Research 2018; 24(23), 6053-6065.
[22] S Hong, C Liow, J Yuk, H Byon, Y Yang, E Cho, J Yeom, G Park, H Kang, S Kim, Y Shim, M Na, C Jeong, G Hwang, H Kim, H Kim, S Eom, S Cho, H Jun, ..., HM Lee. Reducing time to discovery: Materials and molecular modeling, imaging, informatics, and integration. ACS Nano 2021; 15(3), 3971-3995.
[23] R Peach, A Arnaudon and M Barahona. Relative, local and global dimension in complex networks. Nature Communications 2022; 13(1), 3088.
[24] AG Atanasov, SB Zotchev, VM Dirsch and CT Supuran. Natural products in drug discovery: Advances and opportunities. Nature Reviews Drug Discovery 2021; 20(3), 200-216.
[25] FS Li and JK Weng. Demystifying traditional herbal medicine with modern approach. Nature Plants 2017; 3(8), 1-7.
[26] M Verni and F Casanova. The potential of food by-products: Bioprocessing, bioactive compounds extraction and functional ingredients utilization. Foods 2022; 11(24), 4092.
[27] H El-Ramady, P Hajdú, G Törős, K Badgar, X Llanaj, A Kiss, N Abdalla, AE Omara, T Elsakhawy, H Elbasiouny, F Elbehiry, M Amer, ME El-Mahrouk and J Prokisch. Plant nutrition for human health: A pictorial review on plant bioactive compounds for sustainable agriculture. Sustainability 2022; 14(14), 8329.
[28] DJ Newman and GM Cragg. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. Journal of Natural Products 2020; 83(3), 770-803.
[29] T Rodrigues. Harnessing the potential of natural products in drug discovery from a cheminformatics vantage point. Organic & Biomolecular Chemistry 2017; 15(44), 9275-9282.
[30] M Ghasemi, T Turnbull, S Sebastian and I Kempson. The MTT assay: Utility, limitations, pitfalls, and interpretation in bulk and single-cell analysis. International Journal of Molecular Sciences 2021; 22(23), 12827.
[31] Y Song and S Zhang. ComProliM: A cell growth assay robust to initial cell number in co-culture system. Heliyon 2023; 9(9), e19433.
[32] I Falke, FM Troschel, H Palenta, MT Löblein, K Brüggemann, K Borrmann, HT Eich, M Götte and B Greve. Knockdown of the stem cell marker Musashi-1 inhibits endometrial cancer growth and sensitizes cells to radiation. Stem Cell Research & Therapy 2022; 13(1), 212.
[33] P Tanamatayarat, P Limtrakul, S Chunsakaow and C Duangrat. Screening of some rubiaceous plants for cytotoxic activity against cervix carcinoma (kb-3-1) cell line. The Thai Journal of Pharmaceutical Sciences 2003; 27(3), 167-172.
[34] Y Cai, M Prochazkova, YS Kim, C Jiang, J Ma, L Moses, K Martin, V Pham, N Zhang, SL Highfill, RP Somerville, DF Stroncek and P Jin. Assessment and comparison of viability assays for cellular products. Cytotherapy 2024; 26(2), 201-209.
[35] BK Shoichet. Screening in a spirit haunted world. Drug Discovery Today 2006; 11(13-14), 607-615.
[36] JP Hughes, S Rees, SB Kalindjian and KL Philpott. Principles of early drug discovery. British Journal of Pharmacology 2011; 162(6), 1239-1249.
[37] MA Mahmood, AH Abd and EJ Kadhim. Assessing the cytotoxicity of phenolic and terpene fractions extracted from Iraqi Prunus arabica against AMJ13 and SK-GT-4 human cancer cell lines. F1000Research 2024; 12, 433.
[38] M Nafie, I Shawish, S Fahmy, M Diab, M Abdelfattah, B Hassen, K Darwish, A El-Faham and A Barakat. Recent advances in the halogenated spirooxindoles as novel anticancer scaffolds: Chemistry and bioactivity approach. RSC Advances 2025; 15(28), 22336-22375.
[39] M Furqan, FS Wahyuni, M Susanti and D Hamidi. Evaluation of Garcinia cowa leaf extract as a potential anticancer agent: Cytotoxicity, selectivity, and apoptotic effects on MCF-7/HER-2 cells. Tropical Journal of Natural Product Research 2025; 9(2), 846-852.
[40] A Chhikara, PK Roayapalley, H Sakagami, S Amano, K Satoh, Y Uesawa, U Das, S Das, EA Borrego, CD Guerena, CR Hernandez, RJ Aguilera and JR Dimmock. Novel unsymmetric 3, 5-bis (benzylidene)-4-piperidones that display tumor-selective toxicity. Molecules 2022; 27(19), 6718.
[41] ID Zlotnikov, NV Dobryakova, AA Ezhov and EV Kudryashova. Achievement of the selectivity of cytotoxic agents against cancer cells by creation of combined formulation with terpenoid adjuvants as prospects to overcome multidrug resistance. International Journal of Molecular Sciences 2023; 24(9), 8023.
[42] T Mukohara, H Shimada, N Ogasawara, R Wanikawa, M Shimomura, T Nakatsura, G Ishii, JO Park, PA Jänne, N Saijo and H Minami. Sensitivity of breast cancer cell lines to the novel insulin-like growth factor-1 receptor (IGF-1R) inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression. Cancer Letters 2009; 282(1), 14-24.
[43] H Hartog, W Graaf, H Boezen and J Wesseling. Treatment of breast cancer cells by IGF1R tyrosine kinase inhibitor combined with conventional systemic drugs. Anticancer Research 2012; 32(4), 1309-1318.
[44] A Stanley, G Ashrafi, A Seddon and H Modjtahedi. Synergistic effects of various HER inhibitors in combination with IGF1R, C-MET and SRC targeting agents in breast cancer cell lines. Scientific Reports 2017; 7, 3964.
[45] J Adjo Aka and SX Lin. Comparison of functional proteomic analyses of human breast cancer cell lines T47D and MCF7. PLoS One 2012; 7(2), e31532.
[46] T Pantsar, PD Kaiser, M Kudolo, M Forster, U Rothbauer and SA Laufer. Decisive role of water and protein dynamics in residence time of p38α MAP kinase inhibitors. Nature Communications 2022; 13(1), 569.
[47] R Lazim, D Suh and S Choi. Advances in molecular dynamics simulations and enhanced sampling methods for the study of protein systems. International Journal of Molecular Sciences 2020; 21(17), 6339.
[48] S Rathod, P Chavan, D Mahuli, S Rochlani, S Shinde, S Pawar, P Choudhari, R Dhavale, P Mudalkar and F Tamboli. Exploring biogenic chalcones as DprE1 inhibitors for antitubercular activity via in silico approach. Journal of Molecular Modeling 2023; 29(4), 113.
[49] T Wang and Z Wang. Targeting the “Undruggable”: Small-molecule inhibitors of proliferating cell nuclear antigen (PCNA) in the spotlight in cancer therapy. Journal of Medicinal Chemistry 2025; 68(3), 2058-2088.
[50] VK Bhardwaj and R Purohit. A new insight into protein-protein interactions and the effect of conformational alterations in PCNA. International Journal of Biological Macromolecules 2020; 148, 999-1009.
[51] F Li, A Phadte, M Bhatia, S Barndt, A Carlo, CFD Hou, R Yang, S Strock and A Pluciennik. Structural and molecular basis of PCNA-activated FAN1 nuclease function in DNA repair. Nature Communications 2025; 16(1), 4411.
[52] I Ahmad, V Jagatap and H Patel. Application of density functional theory (DFT) and response surface methodology (RSM) in drug discovery, phytochemistry. In: C Egbuna, M Rudrapal and H Tijjani (Eds.). Phytochemistry, computational tools and databases in drug discovery. Elsevier, Amsterdam, Netherlands, 2023.
[53] TN Mohammed Musthafa, K Snigdha, AM Asiri, TR Sobahi and M Asad. Green synthesis of chromonyl chalcone and pyrazoline as potential antimicrobial agents: DFT, molecular docking and antimicrobial studies. Journal of Molecular Structure 2023; 1271, 133993.
[54] M Enneiymy, HA Mohammad-Salim, A Oubella, JV de Julián-Ortiz, HM Tsahnang Fofack, SA Alotaibi, MM Mbake and MYA Itto. In-silico analysis of benzo-selenadiazole hybrids: Reactivity and anticancer potential assessed through DFT, molecular dynamics, molecular docking, and ADMET. Polycyclic Aromatic Compounds 2025; 45(8), 1536-1558.
[55] A Mahal, M Al-Janabi, V Eyüpoğlu, A Alkhouri, S Chtita, MM Kadhim, AJ Obaidullah, JM Alotaibi, X Wei and MRF Pratama. Molecular docking, drug-likeness and DFT study of some modified tetrahydrocurcumins as potential anticancer agents. Saudi Pharmaceutical Journal 2024; 32(1), 101889.
[56] A Das, A Das and BK Banik. Influence of dipole moments on the medicinal activities of diverse organic compounds. Journal of the Indian Chemical Society 2021; 98(2), 100005.
[57] J Darlami and S Sharma. The role of physicochemical and topological parameters in drug design. Frontiers in Drug Discovery 2024; 4, 1424402.
[58] G Ferraro and A Merlino. Investigation of metallodrug/protein interaction by X-ray crystallography and complementary biophysical techniques. Inorganic Chemistry Frontiers 2025; 12(9), 3345-3366.
[59] VM Patil, SP Gupta, N Masand and K Balasubramanian. Experimental and computational models to understand protein-ligand, metal-ligand and metal-DNA interactions pertinent to targeted cancer and other therapies. European Journal of Medicinal Chemistry Reports 2024; 10, 100133.
[60] MD Mellaoui, K Zaki, K Abbiche, A Imjjad, R Boutiddar, A Sbai, A Jmiai, SE Issami, AM Lamsabhi and H Zejli. In silico anticancer activity of isoxazolidine and isoxazolines derivatives: DFT study, ADMET prediction, and molecular docking. Journal of Molecular Structure 2024; 1308, 138330.
[61] P Bhadra, P Yadav, S Kaur, PT Hanumantharayudu and S Arunachalam. The role of ferroptosis in doxorubicin-induced cardiotoxicity - An update. Life Sciences 2025; 380, 123945.